Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 3 by Conner Chen and Version 2 by Conner Chen.
The fluorine atom exhibits many unique properties, including a small atomic radius, large electronegativity, and minimal polarizability. Thus, when coupled with carbon in the form of a C-F bond, organofluorine compounds with highly sought after properties can be obtained. Perfluoropyridine (PFPy) is an organofluorine compound that has been employed for a variety of applications, from straightforward chemical synthesis to more advanced functions, such as fluorinated networks and polymers. This can be directly attributed to the highly reactive nature of PFPy, especially towards nucleophilic aromatic substitution (SNAr).
- perfluoropyridine
1. Introduction
The fluorine atom exhibits many unique properties, including a small atomic radius, large electronegativity, and minimal polarizability [1]. Thus, when coupled with carbon in the form of a C-F bond, organofluorine compounds with highly sought after properties can be obtained [2]. Many of these fluorinated compounds display increased hydrophobicity, as well as enhanced chemical and oxidative resistance, and improved thermal stability [3]. Accordingly, organofluorine compounds have found applications in a range of areas, such as medical imaging [4][5], pharmaceuticals [6], and agrochemicals [7]. Furthermore, driven by the growing demand to develop novel high-performance materials, fluorinated compounds are often used as the building blocks of more complex chemical systems, such as fluoropolymers and fluorinated network materials [8].
There are a number of different organofluorine compounds that have been used as starting materials for more complex systems and polymers. Organofluorine compounds used for these materials can be derived directly from a commercially sourced fluorinated starting material or prepared in-house using selective or direct fluorination techniques [9][10][11]. The motivation for choosing one feedstock over another weighs heavily on the overall synthetic demand of the polymer or material, scalability, cost, and desired properties. With the growing variety of commercially available fluorinated starting materials, often times, researchers will utilize commercial sources versus turning to the more synthetically demanding routes of direct fluorination.
Perfluoropyridine, or PFPy, is one example of these fluorinated compounds that has been used for a wide variety of purposes, ranging from general synthetic reactions [12][13] to complex pharmaceuticals [14][15][16] and materials [17][18][19]. Although it can be prepared in house by employing the methods described below, PFPy is commercially available, and is considered to be an economically viable option in modern times. Furthermore, PFPy is a very reactive molecule and can be used to develop more complex fluorinated starting materials.
2. Discovery and Synthesis of Perfluoropyridine
The first reported synthetic methods for obtaining PFPy occurred in the early 1960s and involved defluorination of perfluoropiperidine [20][21][22][23]. In these instances, perfluoropiperidine, which was synthesized electrochemically from pyridine and anhydrous hydrogen fluoride [24], was passed over a metal, either iron or nickel, at high temperatures. PFPy was then isolated through chromatographic methods. When iron was implemented, PFPy was obtained at a 26% yield. A slightly lower recovery, 12%, was found when defluorination was conducted using nickel (Scheme 1) [20][21][22].
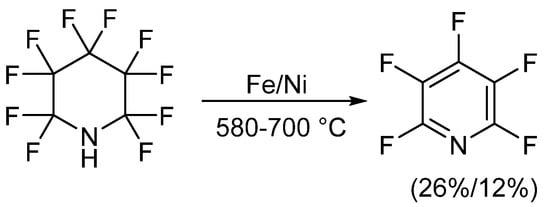
Scheme 1. Synthesis of PFPy from perfluoropiperidine.
In 1964 and 1965, two different groups—Chambers et al. [25][26][27] and Banks et al. [28][29]—published similar methods for the synthesis of PFPy. Here, the authors were able to prepare PFPy by heating pentachloropyridine in an autoclave with anhydrous potassium fluoride. Pentachloropyridine was prepared by reacting pyridine with phosphorous pentachloride [30]. In this instance, a mixture of products was obtained but could be separated by distillation (Scheme 2) [25][26][27][28][29].
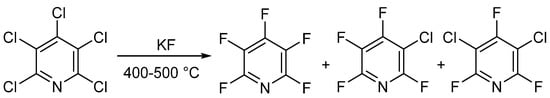
Scheme 2. Synthesis of PFPy using anhydrous potassium fluoride.
The total amount of halogenated products was about 90%, and the ratio of products could be tuned by changing the temperature and reaction time. Under optimal conditions, PFPy could be obtained at a yield of 83% [29]. The method shown in Scheme 2 is still the gold standard for synthesizing PFPy commercially.
Since the development of the commercial method for obtaining PFPy in the mid-1960s, little progress on obtaining PFPy by other means has been made. Nearly 20 years later, Banks et al. obtained trace quantities of PFPy by co-pyrolyzing pentafluoro(trychloromethy)benzene and 4-(dichloroamino)tetrafluoropyridine under nitrogen [31]. The same year, Coe and Sleigh pyrolyzed a number of pyrrolidines in the presence of iron [32]. This reaction produced a mixture of products, and similar to the report by Banks [31], PFPy was obtained in very low quantities (<12%). Later, in 1982, Plevey and co-workers were able to obtain PFPy at a yield of 40% by fluorinating pyridine with cesium tetrafluorocobalatate (III) [33]. Despite this, they were also met with challenges and encountered a point of diminishing returns on the yield when the reaction was attempted above a 5 g scale (Scheme 3) [33].
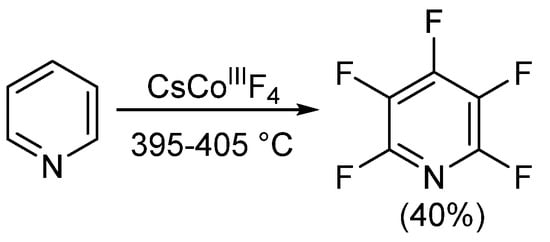
Scheme 3. Synthesis of PFPy using cesium tetrafluorocobalatate (III).
In a final example published in 2004 by Bardin et al., the authors were able to obtain PFPy by the dehalogenation of C5Cl4F5N in the presence of iron or zinc [34]. In all cases, a mixture of products was obtained and non-isolated yields, as determined by GLC analysis, remained average when compared to those obtained by the method presented in Scheme 2 (Table 1).
Table 1.
Reaction summary from Bardin et al.
[34]
.
| Entry | Catalyst/co-Catalyst | Conditions | PFPy Yield (%) |
|---|---|---|---|
| 1 | Fe | 500 °C | 40–43 |
| 2 | Zn | THF, 80 °C, 5 h | 18 |
| 3 | Zn/SOCl2 | Sulfolane, 80 °C, 5 h | 30 |
| 4 | Zn/CuSO4 | Sulfolane, 80 °C, 5 h | 38 |
| 5 | Zn/CuSO4 | Diglyme, 80 °C, 5 h | 29 |
| 6 | Zn/complex NiCl2x6H2O + bpy | DMA, 80 °C, 5 h | 34 |
3. Chemistry of Perfluoropyridine
Since its initial reported synthesis in the early 1960s, there have been over 600 publications in which PFPy has been utilized as a starting material or reagent [35]. With that, the chemical transformations of PFPy are well documented and some noteworthy methodologies include C-F bond activation [36], photoredox reactions [37][38], hydrodefluorination [39][40], and nucleophilic addition [41]. With the variety of readily available nucleophiles, the latter of these known chemical transformations has shown to be quite versatile. Furthermore, PFPy possesses unique regioselectivity, whereby stoichiometric addition to the 4-para-position is exclusive with a broad range of O-, N-, S, and C-nucleophiles under mild basic conditions. Sequential addition to the 2,6-ortho-positions can also be accomplished, dividing substitution at the 3,5-meta-positions (Scheme 4).
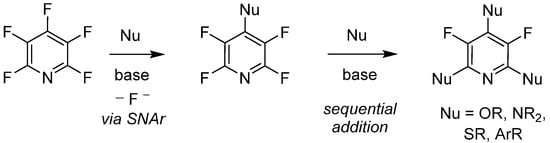
Scheme 4. PFPy as a regio- and chemo-selective nucleophile.
This sequence is commonly accepted to proceed under classic nucleophilic aromatic substitution (SNAr). However, it has been recently debated whether these reactions proceed via a two-step process with a Meisenheimer intermediate or, rather, as a concerted reaction [42]. A concerted nucleophilic aromatic substitution reaction (CSNAr) mechanism supports the notion that electron-poor arenes cannot effectively stabilize a negative charge during Meisenheimer complex formation, but can favorably undergo substitution under mild conditions with a good leaving group, in this case fluoride. Furthermore, PFPy is also chemo-selective with a large pool of di-nucleophiles, which has further expanded the utility of this molecule. A consolidated review of perfluoroaromatic chemistry, in particularly with PFPy, has highlighted comprehensive nucleophilic addition–substitution transformations demonstrating chemo- and regioselectivity [43].
Vlasov et al. has demonstrated that PFPy undergoes reversible substitution with fluoride (F−) under basic conditions with good nucleophiles—in this case, phenols, as shown in Scheme 5 [44]. This work was further expanded to include a DFT modeling study of the reversibility effects of electron-rich/poor aryl ether substitutions at the 2,6-ortho-positions of PFPy [45]. Brittain et al. exploited these phenomena in order to utilize PFPy as a robust protecting group for a diverse pool of phenolics that exhibited mild cleavage/deprotection [12].
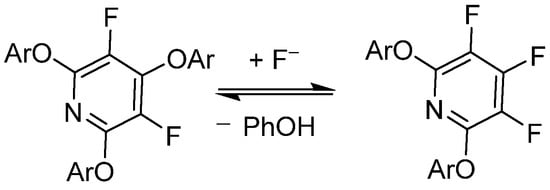
Scheme 5. PFPy and its reversibility to complex products with phenols.
Perfluoropyridine has been employed as a nucleophilic fluorination reagent for both alkyl halides (Scheme 6, right-to-left) [46] as well as deoxyfluorination of carboxylic acids (Scheme 6, left-to-right) [47] for the preparation of a versatile pool of substrates. Fluorination of organo-halides (R–X) is achieved after generation of the air-stable fluoride salt from the addition of PFPy and dimethylaminopyridine [46]. Furthermore, the in situ generation of acyl fluorides is readily accomplished by the nucleophilic addition of carboxylic acids with PFPy [47]. The ester intermediate then undergoes cleavage from unsequestered fluoride, producing phenol as a by-product and the desired acyl fluoride. This strategy was expanded to include the ‘one-pot’ preparation of amides and esters utilizing PFPy as a coupling agent.
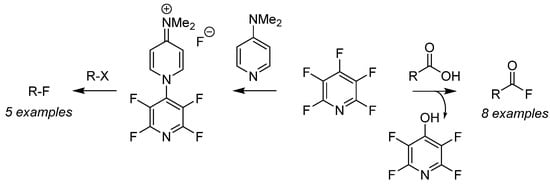
Scheme 6. PFPyas a nucleophilic fluorinating agent.
The last examples of purposeful manipulation of PFPy involve site selective defluorination and halogen exchange (halex), as illustrated in Scheme 7 [48][49]. Both processes generate a regioselective substitution depending on various reaction conditions. Senaweera et al. demonstrated high photocatalytic turnover with low-Ir complex loading for the preparation of deyhdrofluorinated PFPy utilizing a flow reactor design, demonstrating the scalability of such useful intermediates [48]. The halex reverse-substitution was achieved in the 4-para-position of PFPy with LiCl, and additional exchanges on the ring with chlorine were achieved from a combination of kinetic and thermodynamic control [49].
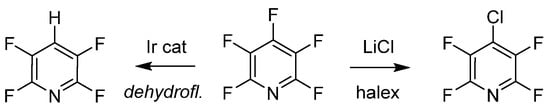
Scheme 7. PFPy as a scaffold for selective hydrodefluorination.
References
- Clark, C.H.D. Atomic radius of fluorine. Nature 1934, 134, 99–100.
- Smart, B.E. Characteristics of the C-F Systems. In Organofluorine Chemistry—Principles and Commercial Applications; Banks, R.E., Smart, B.E., Tatlow, J.C., Eds.; Plenum Press: New York, NY, USA; London, UK, 1994; pp. 57–82. ISBN 978–1-4899-1202-2.
- Tressaud, A. 2—Fluorine, a key element for the 21st century. In Fluorine; Tressaud, A., Ed.; Elsevier: Amsterdam, The Netherlands, 2019; Volume 5, pp. 77–150. ISBN 978-0-12-812990-6.
- Cosco, D.; Fattal, E.; Fresta, M.; Tsapis, N. Perfluorocarbon-loaded micro and nanosystems for medical imaging: A state of the art. J. Fluor. Chem. 2015, 171, 18–26.
- Hequet, E.; Henoumont, C.; Muller, R.N.; Laurent, S. Fluorinated MRI contrast agents and their versatile applications in the biomedical field. Future Med. Chem. 2019, 11, 1157–1175.
- Inoue, M.; Sumii, Y.; Shibata, N. Contribution of Organofluorine Compounds to Pharmaceuticals. ACS Omega 2020, 5, 10633–10640.
- Ogawa, Y.; Tokunaga, E.; Kobayashi, O.; Hirai, K.; Shibata, N. Current Contributions of Organofluorine Compounds to the Agrochemical Industry. iScience 2020, 23, 101467.
- Smith, D.W., Jr.; Iacono, S.T.; Iyer, S.S. (Eds.) Handbook of Fluoropolymer Science and Technology; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2014; ISBN 978-0-470-07993-5.
- Okazoe, T. Overview on the history of organofluorine chemistry from the viewpoint of material industry. Proc. Jpn. Acad. B-Phys. 2009, 85, 276–289.
- Okazoe, T. Development of the “PERFECT” direct fluorination method and its industrial applications. J. Fluor. Chem. 2015, 174, 120–131.
- Daglar, O.; Cakmakci, E.; Gunay, U.S.; Hizal, G.; Tunca, U.; Durmaz, H. A Straightforward Method for Fluorinated Polythioether Synthesis. Macromolecules 2020, 53, 2965–2975.
- Brittain, W.D.G.; Cobb, S.L. Tetrafluoropyridyl (TFP): A general phenol protecting group readily cleaved under mild conditions. Org. Biomol. Chem. 2019, 17, 2110–2115.
- Liljenberg, M.; Brinck, T.; Herschend, B.; Rein, T.; Tomasi, S.; Svensson, M. Predicting Regioselectivity in Nucleophilic Aromatic Substitution. J. Org. Chem. 2012, 77, 3262–3269.
- Darehkordi, A.; Ramezani, M.; Rahmani, F.; Ramezani, M. Design, Synthesis and Evaluation of Antibacterial Effects of a New Class of Piperazinylquinolone Derivatives. J. Heterocycl. Chem. 2016, 53, 89–94.
- Yang, W.-Y.; Breiner, B.; Kovalenko, S.V.; Ben, C.; Singh, M.; LeGrand, S.N.; Sang, Q.-X.A.; Strouse, G.F.; Copland, J.A.; Alabugin, I.V. C-Lysine Conjugates: pH-Controlled Light-Activated Reagents for Efficient Double-Stranded DNA Cleavage with Implications for Cancer Therapy. J. Am. Chem. Soc. 2009, 131, 11458–11470.
- Gimenez, D.; Mooney, C.A.; Dose, A.; Sandford, G.; Coxon, C.R.; Cobb, S.L. The application of perfluoroheteroaromatic reagents in the preparation of modified peptide systems. Org. Biomol. Chem. 2017, 15, 4086–4095.
- Sandford, G. Macrocycles from Perhalogenated Heterocycles. Chem. Eur. J. 2003, 9, 1464–1469.
- Trawny, D.; Kunz, V.; Reissig, H.-U. Modular Syntheses of Star-Shaped Pyridine, Bipyridine, and Terpyridine Derivatives by Employing Sonogashira Reactions. Eur. J. Org. Chem. 2014, 2014, 6295–6302.
- Gutov, A.V.; Rusanov, E.B.; Ryabitskii, A.B.; Chernega, A.N. Octafluoro-4,4′-bipyridine and its derivatives: Synthesis, molecular and crystal structure. J. Fluor. Chem. 2010, 131, 278–281.
- Banks, R.E.; Ginsberg, A.E.; Haszeldine, R.N. Pentafluoropyridine. Proc. Chem. Soc. 1960, 211.
- Burdon, J.; Gilman, D.J.; Patrick, C.R.; Stacey, M.; Tatlow, J.C. Pentafluoropyridine. Nature 1960, 186, 231–232.
- Banks, R.E.; Ginsberg, A.E.; Haszeldine, R.N. 338. Heterocyclic polyfluoro-compounds. Part I. Pentafluoropyridine. J. Chem. Soc. 1961, 1740–1743.
- Haszeldine, R.N.; Banks, R.E.; Ginsberg, A.E. Perfluorinated Aromatic Heterocyclic Compounds. UK Patent GB 980,248, 13 January 1965.
- Haszeldine, R.N.; Nyman, F. Perfluoroalkyl derivatives of sulfur. V. α,α-Difluoro-α-(trifluorothio)acetic acid. J. Chem. Soc. 1956, 2684–2689.
- Chambers, R.D.; Hutchinson, J.; Musgrave, W.K.R. Pentafluoro- and chlorofluoropyridines. Proc. Chem. Soc. 1964, 83.
- Chambers, R.D.; Hutchinson, J.; Musgrave, W.K.R. Fluoropyridines and Their Derivatives. U.S. Patent BE 660, 873, 10 March 1965.
- Chambers, R.D.; Hutchinson, J.; Rodgerson Musgrave, W.K. Fluorinated Pyridines. UK Patent GB 1,107,881, 27 March 1968.
- Banks, R.E.; Haszeldine, R.N.; Latham, J.V.; Young, I.M. Polyfluoropyridines. Chem. Ind. 1964, 20, 835.
- Banks, R.E.; Haszeldine, R.N.; Latham, J.V.; Young, I.M. 95. Heterocyclic polyfluoro-compounds. Part VI. Preparation of pentafluoropyridine and chlorofluoropyridines from pentachloropyridine. J. Chem. Soc. 1965, 594–597.
- Sell, W.J.; Dootson, F.W. XLV.—The chlorine derivatives of pyridine. Part I. J. Chem. Soc. Trans. 1898, 73, 432–441.
- Banks, R.E.; Barlow, M.G.; Mamaghani, M. Vacuum pyrolysis of 4-(dichloroamino)tetrafluoropyridine and related compounds. J. Fluor. Chem. 1981, 17, 197–203.
- Coe, P.L.; Sleigh, J.H. Highly fluorinated heterocycles part XV . The metal catalysed defluorinative rearrangement of polyfluorinated pyrrolidines to polyfluoropyridines. J. Fluor. Chem. 1981, 17, 403–407.
- Plevey, R.G.; Rendell, R.W.; Tatlow, J.C. Fluorinations with complex metal fluorides. Part 6 fluorination of yridine and related compounds with caesium tetrafluorocobaltate(III). J. Fluor. Chem. 1982, 21, 159–169.
- Bardin, V.V.; Trukhin, D.V.; Adonin, N.Y.; Starichenko, V.F. Dehalogenative aromatization of perchlorofluoroalicyclic compounds C6Cl6F6, C10Cl8F8 and C5Cl4F5N in the vapour phase or in solution. J. Fluor. Chem. 2004, 125, 1431–1435.
- SciFinder. Available online: https://scifinder.cas.org/scifinder (accessed on 28 November 2021).
- Amii, H.; Uneyama, K. C–F Bond Activation in Organic Synthesis. Chem. Rev. 2009, 109, 2119–2183.
- Lipilin, D.L.; Frumkin, A.E.; Tyurin, A.Y.; Levin, V.V.; Dilman, A.D. Photoredox Catalyzed Dealkylative Aromatic Halogen Substitution with Tertiary Amines. Molecules 2021, 26, 3323.
- Zubkov, M.O.; Kosobokov, M.D.; Levin, V.V.; Kokorekin, V.A.; Korlyukov, A.A.; Hu, J.; Dilman, A.D. A novel photoredox-active group for the generation of fluorinated radicals from difluorostyrenes. Chem. Sci. 2020, 11, 737–741.
- McKay, D.; Riddlestone, I.M.; Macgregor, S.A.; Mahon, M.F.; Whittlesey, M.K. Mechanistic Study of Ru-NHC-Catalyzed Hydrodefluorination of Fluoropyridines: The Influence of the NHC on the Regioselectivity of C–F Activation and Chemoselectivity of C–F versus C–H Bond Cleavage. ACS Catal. 2015, 5, 776–787.
- Arévalo, A.; Tlahuext-Aca, A.; Flores-Alamo, M.; García, J.J. On the Catalytic Hydrodefluorination of Fluoroaromatics Using Nickel Complexes: The True Role of the Phosphine. J. Am. Chem. Soc. 2014, 136, 4634–4639.
- Wielgat, J.; Domagala, Z. Reaction of pentafluoropyridine with bifunctional nucleophiles. Rocz. Chem. 1975, 49, 1039–1041.
- Neumann, C.N.; Hooker, J.M.; Ritter, T. Concerted nucleophilic aromatic substitution with 19F− and 18F−. Nature 2016, 534, 369–373.
- Sandford, G. Perfluoroheteroaromatic Chemistry: Multifunctional Systems from Perfluorinated Heterocycles by Nucleophilic Aromatic Substitution Processes. In Halogenated Heterocycles: Synthesis Applications and Environment; Iskra, J., Ed.; Springer: Berlin/Heidelberg, Germany, 2012; pp. 1–31. ISBN 978-3-642-43950-6.
- Vlasov, V.M.; Aksenov, V.V.; Rodionov, P.P.; Beregovaya, I.V.; Shchegoleva, L.N. Unusual Lability of Pentafluorophenoxy Group in Reactions of Potassium Aroxides with Pentafluoropyridine. Russ. J. Org. Chem. 2002, 38, 115–125.
- Fuhrer, T.J.; Houck, M.; Corley, C.A.; Iacono, S.T. Theoretical Explanation of Reaction Site Selectivity in the Addition of a Phenoxy Group to Perfluoropyridine. J. Phys. Chem. A 2019, 123, 9450–9455.
- Murray, C.B.; Sandford, G.; Korn, S.R.; Yufit, D.S.; Howard, J.A.K. New fluoride ion reagent from pentafluoropyridine. J. Fluor. Chem. 2005, 126, 569–574.
- Brittain, W.D.G.; Cobb, S.L. Carboxylic Acid Deoxyfluorination and One-Pot Amide Bond Formation Using Pentafluoropyridine (PFP). Org. Lett. 2021, 23, 5793–5798.
- Senaweera, S.M.; Singh, A.; Weaver, J.D. Photocatalytic Hydrodefluorination: Facile Access to Partially Fluorinated Aromatics. J. Am. Chem. Soc. 2014, 136, 3002–3005.
- Froese, R.D.J.; Whiteker, G.T.; Peterson, T.H.; Arriola, D.J.; Renga, J.M.; Shearer, J.W. Computational and Experimental Studies of Regioselective SNAr Halide Exchange (Halex) Reactions of Pentachloropyridine. J. Org. Chem. 2016, 81, 10672–10682.
More