Apoptosis is an evolutionarily conserved and tightly regulated cell death pathway. Physiological cell death is important for maintaining homeostasis and optimal biological conditions by continuous elimination of undesired or superfluous cells. The BH3-only pro-apoptotic members are strong inducers of apoptosis. The pro-apoptotic BH3-only protein Noxa activates multiple death pathways by inhibiting the anti-apoptotic B-cl-2 ell lymphoma-2 (Bcl-2) family protein, Mcl-1, and other protein members leading to Bax and Bak activation and MOMP mitochondrial outer membrane permeabilization (MOMP). On the other hand, Puma is induced by p53-dependent and p53-independent apoptotic stimuli in several cancer cell lines. Moreover, this protein is involved in several physiological and pathological processes, such as immunity, cancer, and neurodegenerative diseases. Future heat shock research could disclose the effect of hyperthermia on both Noxa and BH3-only proteins. This suggests post-transcriptional mechanisms controlling the translation of both Puma and Noxa mRNA in heat-shocked cells.
- Noxa
- Puma
- Bcl-2 proteins
- p53
- heat-shock
- caspase
- ion channels
- apoptosis
1. Introduction
2. Bcl-2 Proteins and the Regulation of Mitochondrial Outer Membrane Permeability
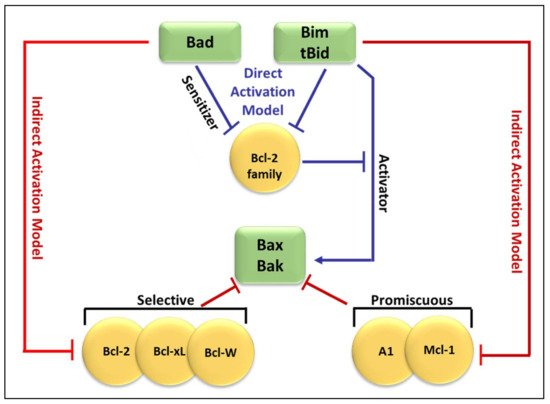
Molecular insights into apoptosis first emerged during the 1980s and 1990s from a convergence of mammalian cancer cytogenetics and genetic studies on developmentally programmed cell death in Caenorhabditis elegans [38][35]. The previously unknown gene Bcl-2 was identified from the breakpoint region of a recurrent chromosomal translocation (18q21) in human follicular lymphoma [7,39][7][36]. The Bcl-2 family of proteins are central regulators of stress-induced apoptosis as they control diverse survival and death signals that are generated inside and outside the cell [40,41][37][38]. Bcl-2 proteins are found in all cells and consist of 12 distinct members, many of which are expressed as alternate isoforms. Structurally, they all contain at least one of the conserved sequence motifs called Bcl-2 BH domains. This family is functionally subdivided into two classes based on their activity and number of BH domains: anti-apoptotic and pro-apoptotic proteins including the BH3-only members [19,41,42][16][38][39]. The mutual interaction between pro-apoptotic and anti-apoptotic members establishes the threshold that determines whether a cell should survive or die [43][40]. In a sense, they act as checkpoints through which survival and death signals must pass to elicit the cell’s fate as they control stress-induced cell death [44][41]. The anti-apoptotic Bcl-2 members, including Bcl-2 itself, Bcl-xL, Mcl-1, Bcl-W, and A1/BFL-1 share four conserved BH domains of structural homology. These proteins prevent cell death against diverse cytotoxic signals, both physiological and imposed. This maintains the integrity of the ER, mitochondrial, and nuclear membranes, thus protecting cells from apoptosis [45,46][42][43]. The proximity of BH 1, 2, and 3 form a hydrophobic pocket that operates as a receptor for the BH3 domain of the pro-apoptotic BH3-only members. The anti-apoptotic Bcl-2 proteins work together to inhibit the release of cytochrome c from the mitochondria by preventing the activation of the pro-apoptotic members Bax and Bak in the OMM [44][41]. The anti-apoptotic proteins’ normal function is to prevent inappropriate cell death and therefore requires the neutralization of the pro-apoptotic proteins by the anti-apoptotic members [47][44]. Just as the anti-apoptotic Bcl-2 proteins promote tumourigenesis when deregulated, the pro-apoptotic members function as tumor suppressors. The pro-apoptotic Bcl-2 members are divided into multi-domain effector proteins, such as Bax, Bak, and the less well-known Bok, as well as the large subgroup of BH3-only proteins, all of which trigger or sensitize the cell to apoptosis [48,49,50][45][46][47]. The pro-apoptotic effector proteins Bax, Bak, and Bok possess three BH domains and adopt similar globular structures: a helical bundle surrounding a central hydrophobic core helix [44,51,52][41][48][49]. This groove constitutes a crucial surface for interactions with the BH3 domain of pro-apoptotic members of the Bcl-2 family [38][35]. These interactions primarily occur on intracellular membranes, such as that of the OMM, where many of the Bcl-2 family members are directed by their carboxy-terminal hydrophobic TM domain [53][50]. Unlike the anti-apoptotic members, the active conformation of Bax/Bak damages rather than protects the OMM of stressed cells. This yields the formation of pores leading to membrane dysfunction [54][51]. The most studied pro-apoptotic proteins Bax and Bak exist in an inactive monomeric state in healthy cells. In stressed cells, they are major inducers of mitochondrial outer membrane permeabilization (MOMP) [55][52]. Recent studies have focused on the effects of the Bcl-2 family members on MOMP. Although over a decade has passed since the discovery of the BH3-only protein Puma, the question of how this protein activates Bax/Bak, consequently leading to MOMP, remains unresolved. At present, two mechanisms have been proposed concerning the relationship of the Bcl-2 protein family members employing direct or indirect activation of the mitochondrial apoptotic pathway, shown in Figure 21 [44,47,54][41][44][51]. Two models are explaining Bak/Bax activation through the interaction of the Bcl-2 protein family members: the direct activation model and the indirect activation model.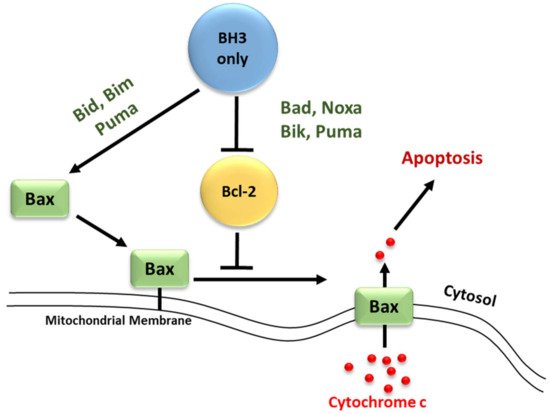
3. Regulation of Apoptosis
3.1. Regulators of Cell Death: Caspases
Caspases (Cysteinyl aspartate-specific proteases) are a family of proteins activated by a variety of stimuli representing a vital step for the induction of apoptosis. These proteases initiate and control the cellular death pathway by cleaving a diverse set of cellular proteins [68][57].
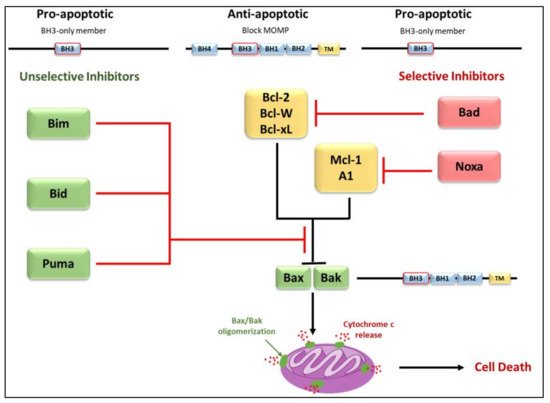
3.2. Regulators of Cell Death: Bcl-2 Family Proteins
3.3. Elucidation of the Caspases Reaction Mechanism
Caspases stand for Cysteine (Cys)-dependent aspartate-directed proteases are a sub-branch of the enzymes family proteases which possess an indispensable role in apoptosis. The commonly proposed mechanism is divided into two principal parts. It describes the cleavage of the peptide bond (amide function).
-
Once, again the alkaline property of one of the nitrogen atoms in the imidazolic part of His deprotonates a water molecule.
-
This deprotonation contributes to the formation of a hydroxide. The strong alkaline and nucleophilic property of the hydroxide contributes to the attack of the electrophilic site of the carbonyl function. This yields a second tetrahedral intermediate.
-
α-thio protonation: Similarly, to the third step of phase 1, the sulfur in I2 constitutes a good leaving group. This enhances the possibility of protonation of the α-thio
-
Formation of the carboxylic acid by regeneration of His and Cys counterparts.
-
The already formed hydrogen bond between His and the first water molecule will favor the deprotonation of the latter. The yielded hydroxide attacks the acyl-enzyme complex on the carbonyl moiety.
-
The yielded alkoxide in I3 will attack the proton already captured by the His part in (a). This will form a germinal diol (I4; Figure 64).
- moiety by the previously protonated nitrogen of the His residue.
- The carboxylate function of the side-chain aspartate will attack one of the hydrogens of the diol. The thiol, acting as a good leaving group, will enhance the possibility of the formation of a carbonyl bond; thereby a carboxylic acid and a thiolate (I5; Figure 64). The computational investigation shows that for the attack of the water molecule, a free energy barrier of about 19 ± 4 kcal/mol must be overcome, these trends are following the experimental results of Sulpizi et al. [88][64].
- Nucleophilic activation: the alkaline property of one of the nitrogen atoms, in the imidazolic part of Histidine (His), deprotonates the hydrogen in the thiol (-SH) group of the Cys residue, yielding a thiolate.
-
Thiolate nucleophilic attack on carbonyl: the carbonyl group of the aspartic peptide bond undergoes a nucleophilic attack by the yielded thiolate in the latter step. This contributes to the formation of a first tetrahedral intermediate.
-
α-amino protonation: the amine group of I1 constitutes a good leaving group. This will enhance the possibility of the protonation of the α-amino moiety by the previous protonated nitrogen of the His residue.
-
Formation of the covalent adduct: the acyl-enzyme complex and the cleavage of the peptide bond.
4. BH3-Only Protein Noxa
4.1. Discovery
4.2. General Features and Transcript Variants
4.3. Regulation of Noxa Expression and Post-Translational Modification
Early observations indicated that Noxa transcription was primarily induced by p53. In wild-type and IRF-1-deficient MEFs, X-ray irradiation caused rapid induction of Noxa mRNA, whereas no Noxa induction was observed in p53¯/¯ MEFs [95][68]. Analysis of the Noxa promoter region revealed a bona fide p53-response element 195 bp upstream of the transcription start site [95,96][68][69]. Additional studies have investigated p53-dependent transcription of Noxa either by in situ hybridization in p53¯/¯ mice or treatment of multiple cell lines with various chemical compounds [98,99,100,101][71][72][73][74]. The p53 independent regulation of Noxa by various stimuli has also been observed. Noxa induction was observed when multiple p53¯/¯ melanoma cell lines, along with PC-3 prostate cells and Saos-2 osteosarcoma cells (both p53 null cell lines) were treated with the γ-secretase inhibitor GSI [102][75]. Hypoxia-induced HIF-1α has been shown to induce Noxa mRNA and protein expression in H719 and Saos-2 cells independent of p53 by binding to a hypoxia response element (HRE), at −1275 bp, within the Noxa promoter [60][76]. Overexpression of adenovirus E1A protein, in the neuroblastoma cell line SH-SY5Y (non-functional p53) and SaOS-2 cells, results in activation of p73 and induction of Noxa mRNA [103][77]. H2O2-induces activating transcription factor 4 can induce Noxa mRNA expression in Jurkat cells by binding to a cAMP response element-binding site within the Noxa promoter [104][78]. A FoxO-binding site has been identified within the Noxa promoter by treatment of Jurkat cells with α-tocopheryl succinate, resulting in activation of FoxO1 and FoxO1-mediated transcription of Noxa [105][79]. Additional studies have been completed that have investigated p53-independent regulation of Noxa transcription [106,107,108,109,110,111,112][80][81][82][83][84][85][86]. In addition to transcriptional regulation of Noxa, proteasomal degradation has been implicated in the control of Noxa protein stability. Noxa has a short half-life [97][70], although it does not contain any PEST or known E3-ligase binding domains. KLF6-SV1 was observed to bind to Noxa and lead to its HDM2-mediated proteasomal degradation upon KLF6-SV1 overexpression in SKOV3 cells [113][87]. Additionally, proteasome inhibition in SKOV3 cells by MG132 causes an increase in both KLF6-SV1 and Noxa [113][87]. Treatment of MM.1S cells with the novel proteasome inhibitor MLN2238 results in increased expression of both p53 and Noxa [114][88]. Treatment with the proteasome inhibitor bortezomib (PS-441, Velcade) results in Noxa mRNA and protein induction in both p53 wild-type and p53-null melanoma cells, but not in normal melanocytes [115][89]. This effect of bortezomib was also observed in vivo and multiple other cell lines [116][90]. A cellular myelocytomatosis viral oncogene (c-MYC) binding site within the Noxa promoter has been identified, and siRNA knockdown of c-MYC reduced bortezomib-induced Noxa mRNA expression in multiple melanoma cell lines, MDA-MB-231 cells, and HeLa cells [117,118][91][92]. Treatment of LX-2 cells (human hepatic stellate cells) with MG132 resulted in increased expression of both Noxa mRNA and Noxa protein, and the MG132-induced apoptosis in LX-2 cells was shown to require Noxa [119][93].4.4. Subcellular Localization and Association with Bcl-2-like Proteins
Assessment by immunostaining has shown that overexpressed mouse Noxa preferentially localizes to the mitochondria and that mutations within either the BH3 domain or MTD prevent mitochondrial localization [95,96][68][69]. Furthermore, Noxa constructs that are missing either the BH3 domain or MTD fail to induce apoptosis [96][69]. This suggests that both the BH3 domain and MTD are required for Noxa-induced apoptosis due to the proximity of the BH3 domain and MTD mutations in either domain may change the overall conformation of Noxa and impair Mcl-1 binding [94][67]. Noxa contains only a single BH3 domain, placing it in the growing category of pro-apoptotic BH3-only proteins. Studies have shown that overexpression of Noxa can significantly induces apoptosis in various cell lines [95[68][69][90],96,116], as well as correlates with MOMP, reactive oxygen species (ROS) accumulation, and cytochrome c release [92,96,125][94][69][95]. The specificity of Noxa towards Mcl-1 and A1 is dependent on key amino acid residues within the Noxa BH3 domain. Mutations in the Noxa BH3 domain (m3) allow Noxa to bind to Bcl-xL with a 100-fold increased affinity versus wild-type Noxa and is a more potent inducer of apoptosis [67][56], while other mutations within the BH3 domain rendered Noxa inactive [95,96][68][69]. Additionally, Bims chimeras containing the Noxa BH3 domain showed reduced apoptotic ability, as compared to the wild-type Bims, which have a high apoptotic potential. Bims-Noxa BH3 chimeras were also restricted to binding Mcl-1, further demonstrating how the Noxa BH3 domain controls binding specificity and apoptotic potential of Noxa [67][56]. Interaction of Noxa with Mcl-1 has also been observed in melanoma cells treated with bortezomib [135][96], and in MDN and Jurkat cells where endogenous Noxa/Mcl-1 complexes were detected [106,136][80][97]. Due to the specificity of Noxa for both Mcl-1 and A1 [67][56], the cellular levels of Mcl-1 and A1 control sensitivity to Noxa-induced apoptosis. Overexpression of Noxa in MEFs leads to Mcl-1 degradation without significant induction of apoptosis [66][55]. Consistent with the idea that Noxa needs to be complemented by Bad, which targets Bcl-xL, Bcl-2, and Bcl-w, to induce apoptosis [67][56], overexpression of both Noxa and Bad was shown to induce apoptosis in MEFs [66,133][55][98]. Overexpression of Noxa in Bcl-xL¯/¯ MEFs induced Bak-dependent apoptosis, demonstrating that Mcl-1 and Bcl-xL constrain Bak and that Noxa specifically engages Mcl-1 to promote Bak-dependent apoptosis [66][55]. Overexpression of Noxa has also been shown to disrupt Mcl-1/Bak complexes in multiple myeloma and B-cell lymphomas and in Jurkat cells [140][99]. Noxa has also been shown to disrupt Mcl-1/Bim complexes in bortezomib-treated MDN cells [136][97].5. BH3-Only Protein Puma
Puma, a p53 Up-regulated Modulator of apoptosis protein, was first discovered and cloned as a transcriptional target of p53 by two independent laboratories 19 years ago [142][100]. In the same year, Han and colleagues identified the bbc3 (Bcl-2 binding component 3) gene that corresponds to the Puma cDNA [143][101]. Puma is a highly efficient pro-apoptotic protein, thought to be one of the most powerful and effective “killers” among the BH3-only proteins. The bbc3 gene has been reported to encode 4 different forms (α, β, γ, and δ) of which only the α and β forms contain the BH3 domain and thus display the pro-apoptotic activity. The length of the α Puma transcript is 1.6–1.9 kb encoding a 193 amino acid protein [142][100]. This protein is highly conserved among vertebrate species, yet shows no significant homologies to any other known proteins aside from those with the BH3 domain [52][49].5.1. Regulation of the BH3-Only Protein Puma
5.2. p53-Dependent Apoptosis
5.3. p53-Independent Apoptosis
References
- Cui, Z.-G.; Piao, J.-L.; Kondo, T.; Ogawa, R.; Tsuneyama, K.; Zhao, Q.-L.; Feril, L.B., Jr.; Inadera, H. Molecular Mechanisms of Hyperthermia-Induced Apoptosis Enhanced by Docosahexaenoic Acid: Implication for Cancer Therapy. Chem. Biol. Interact. 2014, 215, 46–53.
- Kerr, J.F. A Histochemical Study of Hypertrophy and Ischaemic Injury of Rat Liver with Special Reference to Changes in Lysosomes. J. Pathol. Bacteriol. 1965, 90, 419–435.
- Kerr, J.F.; Wyllie, A.H.; Currie, A.R. Apoptosis: A Basic Biological Phenomenon with Wideranging Implications in Tissue Kinetics. Br. J. Cancer 1972, 26, 239–257.
- Taylor, R.C.; Cullen, S.P.; Martin, S.J. Apoptosis: Controlled Demolition at the Cellular Level. Nat. Rev. Mol. Cell Biol. 2008, 9, 231–241.
- Kihlmark, M.; Imreh, G.; Hallberg, E. Sequential Degradation of Proteins from the Nuclear Envelope during Apoptosis. J. Cell Sci. 2001, 114, 3643–3653.
- Yoo, H.; Cha, H.J.; Lee, J.; Yu, E.-O.; Bae, S.; Jung, J.H.; Sohn, I.; Lee, S.-J.; Yang, K.-H.; Woo, S.-H. Specific Proteolysis of the A-Kinase-Anchoring Protein 149 at the Asp582 Residue by Caspases during Apoptosis. Oncol. Rep. 2008, 19, 1577–1582.
- Vaux, D.L.; Cory, S.; Adams, J.M. Bcl-2 Gene Promotes Haemopoietic Cell Survival and Cooperates with c-Myc to Immortalize Pre-B Cells. Nature 1988, 335, 440–442.
- Hanahan, D.; Weinberg, R.A. The Hallmarks of Cancer. Cell 2000, 100, 57–70.
- Liadis, N.; Murakami, K.; Eweida, M.; Elford, A.R.; Sheu, L.; Gaisano, H.Y.; Hakem, R.; Ohashi, P.S.; Woo, M. Caspase-3-Dependent β-Cell Apoptosis in the Initiation of Autoimmune Diabetes Mellitus. Mol. Cell. Biol. 2005, 25, 3620–3629.
- Rohn, T.T.; Head, E. Caspases as Therapeutic Targets in Alzheimer’s Disease: Is It Time to “Cut” to the Chase? Int. J. Clin. Exp. Pathol. 2009, 2, 108.
- Soto-Mercado, V.; Mendivil-Perez, M.; Velez-Pardo, C.; Jimenez-Del-Rio, M. (−)-Epigallocatechin-3-Gallate Diminishes Intra-and Extracellular Amyloid-Induced Cytotoxic Effects on Cholinergic-like Neurons from Familial Alzheimer’s Disease PSEN1 E280A. Biomolecules 2021, 11, 1845.
- Townsend, P.A.; Kozhevnikova, M.V.; Cexus, O.N.; Zamyatnin, A.A.; Soond, S.M. BH3-Mimetics: Recent Developments in Cancer Therapy. J. Exp. Clin. Cancer Res. 2021, 40, 355.
- Tsukada, M.; Ohsumi, Y. Isolation and Characterization of Autophagy-Defective Mutants of Saccharomyces Cerevisiae. FEBS Lett. 1993, 333, 169–174.
- Levine, B.; Klionsky, D.J. Development by Self-Digestion: Molecular Mechanisms and Biological Functions of Autophagy. Dev. Cell 2004, 6, 463–477.
- Sinha, S.; Levine, B. The Autophagy Effector Beclin 1: A Novel BH3-Only Protein. Oncogene 2008, 27, S137–S148.
- Delgado, Y.; Torres, A.; Milián, M. Apoptosis’ Activation Associated to BH3 Only Domain and BCL-2 Homology Domain Proteins: New Way to Design Anti-Cancer Drugs. J. Cancer Prev. Curr. Res. 2019, 10, 54–59.
- D’Orsi, B.; Mateyka, J.; Prehn, J.H. Control of Mitochondrial Physiology and Cell Death by the Bcl-2 Family Proteins Bax and Bok. Neurochem. Int. 2017, 109, 162–170.
- Singh, R.; Letai, A.; Sarosiek, K. Regulation of Apoptosis in Health and Disease: The Balancing Act of BCL-2 Family Proteins. Nat. Rev. Mol. Cell Biol. 2019, 20, 175–193.
- Hatok, J.; Racay, P. Bcl-2 Family Proteins: Master Regulators of Cell Survival. Biomol. Concepts 2016, 7, 259–270.
- Opferman, J.T.; Kothari, A. Anti-Apoptotic BCL-2 Family Members in Development. Cell Death Differ. 2018, 25, 37–45.
- Greaves, G.; Milani, M.; Butterworth, M.; Carter, R.J.; Byrne, D.P.; Eyers, P.A.; Luo, X.; Cohen, G.M.; Varadarajan, S. BH3-Only Proteins Are Dispensable for Apoptosis Induced by Pharmacological Inhibition of Both MCL-1 and BCL-X L. Cell Death Differ. 2019, 26, 1037–1047.
- Huang, K.; O’Neill, K.L.; Li, J.; Zhou, W.; Han, N.; Pang, X.; Wu, W.; Struble, L.; Borgstahl, G.; Liu, Z. BH3-Only Proteins Target BCL-XL/MCL-1, Not BAX/BAK, to Initiate Apoptosis. Cell Res. 2019, 29, 942–952.
- Glab, J.A.; Mbogo, G.W.; Puthalakath, H. BH3-Only Proteins in Health and Disease. In International Review of Cell and Molecular Biology; Elsevier: Oxford, UK, 2017; Volume 328, pp. 163–196.
- Happo, L.; Strasser, A.; Cory, S. BH3-Only Proteins in Apoptosis at a Glance. J. Cell Sci. 2012, 125, 1081–1087.
- Dai, H.; Ding, H.; Peterson, K.L.; Meng, X.W.; Schneider, P.A.; Knorr, K.L.; Kaufmann, S.H. Measurement of BH3-Only Protein Tolerance. Cell Death Differ. 2018, 25, 282–293.
- Ludwig, L.M.; Roach, L.E.; Fisher, J.K.; Walensky, L.D.; LaBelle, J.L. Regulation of Immune Homeostasis by Direct Activator BH3-Only Proteins; AACR: New Orleans, LA, USA, 2016.
- Chen, A.; Madu, C.O.; Lu, Y. The Functional Role of Bcl-2 Family of Proteins in the Immune System and Cancer. Oncomedicine 2019, 4, 17–26.
- Guikema, J.E.; Amiot, M.; Eldering, E. Exploiting the Pro-Apoptotic Function of NOXA as a Therapeutic Modality in Cancer. Expert Opin. Ther. Targets 2017, 21, 767–779.
- Shukla, S.; Saxena, S.; Singh, B.K.; Kakkar, P. BH3-Only Protein BIM: An Emerging Target in Chemotherapy. Eur. J. Cell Biol. 2017, 96, 728–738.
- Warren, C.F.; Wong-Brown, M.W.; Bowden, N.A. BCL-2 Family Isoforms in Apoptosis and Cancer. Cell Death Dis. 2019, 10, 177.
- Lin, R.-W.; Ho, C.-J.; Chen, H.-W.; Pao, Y.-H.; Chen, L.-E.; Yang, M.-C.; Huang, S.-B.; Wang, S.; Chen, C.-H.; Wang, C. P53 Enhances Apoptosis Induced by Doxorubicin Only under Conditions of Severe DNA Damage. Cell Cycle 2018, 17, 2175–2186.
- Wu, Y.; Chen, M.; Jiang, J. Mitochondrial Dysfunction in Neurodegenerative Diseases and Drug Targets via Apoptotic Signaling. Mitochondrion 2019, 49, 35–45.
- Cowan, K.; Anichtchik, O.; Luo, S. Mitochondrial Integrity in Neurodegeneration. CNS Neurosci. Ther. 2019, 25, 825–836.
- Wang, J.; Thomas, H.R.; Li, Z.; Yeo, N.C.F.; Scott, H.E.; Dang, N.; Hossain, M.I.; Andrabi, S.A.; Parant, J.M. Puma, Noxa, P53, and P63 Differentially Mediate Stress Pathway Induced Apoptosis. Cell Death Dis. 2021, 12, 659.
- Czabotar, P.E.; Lessene, G.; Strasser, A.; Adams, J.M. Control of Apoptosis by the BCL-2 Protein Family: Implications for Physiology and Therapy. Nat. Rev. Mol. Cell Biol. 2014, 15, 49–63.
- Tsujimoto, Y.; Cossman, J.; Jaffe, E.; Croce, C.M. Involvement of the Bcl-2 Gene in Human Follicular Lymphoma. Science 1985, 228, 1440–1443.
- Strasser, A.; O’Connor, L.; Dixit, V.M. Apoptosis Signaling. Annu. Rev. Biochem. 2000, 69, 217–245.
- Adams, J.M.; Cory, S. Life-or-Death Decisions by the Bcl-2 Protein Family. Trends Biochem. Sci. 2001, 26, 61–66.
- Roufayel, R. Regulation of Stressed-Induced Cell Death by the Bcl-2 Family of Apoptotic Proteins. Mol. Membr. Biol. 2016, 33, 89–99.
- Borner, C. The Bcl-2 Protein Family: Sensors and Checkpoints for Life-or-Death Decisions. Mol. Immunol. 2003, 39, 615–647.
- Chipuk, J.E.; Moldoveanu, T.; Llambi, F.; Parsons, M.J.; Green, D.R. The BCL-2 Family Reunion. Mol. Cell 2010, 37, 299–310.
- Er, E.; Oliver, L.; Cartron, P.-F.; Juin, P.; Manon, S.; Vallette, F.M. Mitochondria as the Target of the Pro-Apoptotic Protein Bax. Biochim. Biophys. Acta (BBA)-Bioenerg. 2006, 1757, 1301–1311.
- Youle, R.J.; Strasser, A. The BCL-2 Protein Family: Opposing Activities That Mediate Cell Death. Nat. Rev. Mol. Cell Biol. 2008, 9, 47–59.
- Adams, J.M.; Cory, S. The Bcl-2 Apoptotic Switch in Cancer Development and Therapy. Oncogene 2007, 26, 1324–1337.
- Huang, D.C.; Strasser, A. BH3-Only Proteins—Essential Initiators of Apoptotic Cell Death. Cell 2000, 103, 839–842.
- Antonsson, B. Bax and Other Pro-Apoptotic Bcl-2 Family “Killer-Proteins” and Their Victim the Mitochondrion. Cell Tissue Res. 2001, 306, 347–361.
- Giam, M.; Huang, D.C.S.; Bouillet, P. BH3-Only Proteins and Their Roles in Programmed Cell Death. Oncogene 2008, 27, S128–S136.
- Muchmore, S.W.; Sattler, M.; Liang, H.; Meadows, R.P.; Harlan, J.E.; Yoon, H.S.; Nettesheim, D.; Chang, B.S.; Thompson, C.B.; Wong, S.-L. X-Ray and NMR Structure of Human Bcl-x L, an Inhibitor of Programmed Cell Death. Nature 1996, 381, 335–341.
- Hikisz, P.; Kiliańska, Z. PUMA, a Critical Mediator of Cell Death—One Decade on from Its Discovery. Cell. Mol. Biol. Lett. 2012, 17, 646–669.
- Kvansakul, M.; Yang, H.; Fairlie, W.D.; Czabotar, P.E.; Fischer, S.F.; Perugini, M.A.; Huang, D.C.S.; Colman, P.M. Vaccinia Virus Anti-Apoptotic F1L Is a Novel Bcl-2-like Domain-Swapped Dimer That Binds a Highly Selective Subset of BH3-Containing Death Ligands. Cell Death Differ. 2008, 15, 1564–1571.
- Strasser, A.; Cory, S.; Adams, J.M. Deciphering the Rules of Programmed Cell Death to Improve Therapy of Cancer and Other Diseases. EMBO J. 2011, 30, 3667–3683.
- Antignani, A.; Youle, R.J. How Do Bax and Bak Lead to Permeabilization of the Outer Mitochondrial Membrane? Curr. Opin. Cell Biol. 2006, 18, 685–689.
- Kuwana, T.; Bouchier-Hayes, L.; Chipuk, J.E.; Bonzon, C.; Sullivan, B.A.; Green, D.R.; Newmeyer, D.D. BH3 Domains of BH3-Only Proteins Differentially Regulate Bax-Mediated Mitochondrial Membrane Permeabilization Both Directly and Indirectly. Mol. Cell 2005, 17, 525–535.
- Willis, S.N.; Fletcher, J.I.; Kaufmann, T.; van Delft, M.F.; Chen, L.; Czabotar, P.E.; Ierino, H.; Lee, E.F.; Fairlie, W.D.; Bouillet, P. Apoptosis Initiated When BH3 Ligands Engage Multiple Bcl-2 Homologs, Not Bax or Bak. Science 2007, 315, 856–859.
- Willis, S.N.; Chen, L.; Dewson, G.; Wei, A.; Naik, E.; Fletcher, J.I.; Adams, J.M.; Huang, D.C. Proapoptotic Bak Is Sequestered by Mcl-1 and Bcl-XL, but Not Bcl-2, until Displaced by BH3-Only Proteins. Genes Dev. 2005, 19, 1294–1305.
- Chen, L.; Willis, S.N.; Wei, A.; Smith, B.J.; Fletcher, J.I.; Hinds, M.G.; Colman, P.M.; Day, C.L.; Adams, J.M.; Huang, D.C. Differential Targeting of Prosurvival Bcl-2 Proteins by Their BH3-Only Ligands Allows Complementary Apoptotic Function. Mol. Cell 2005, 17, 393–403.
- Crawford, E.D.; Wells, J.A. Caspase Substrates and Cellular Remodeling. Annu. Rev. Biochem. 2011, 80, 1055–1087.
- Fulda, S.; Debatin, K.-M. Extrinsic versus Intrinsic Apoptosis Pathways in Anticancer Chemotherapy. Oncogene 2006, 25, 4798–4811.
- Renault, T.T.; Chipuk, J.E. Death upon a Kiss: Mitochondrial Outer Membrane Composition and Organelle Communication Govern Sensitivity to BAK/BAX-Dependent Apoptosis. Chem. Biol. 2014, 21, 114–123.
- Green, D.R.; Evan, G.I. A Matter of Life and Death. Cancer Cell 2002, 1, 19–30.
- Hüttemann, M.; Pecina, P.; Rainbolt, M.; Sanderson, T.H.; Kagan, V.E.; Samavati, L.; Doan, J.W.; Lee, I. The Multiple Functions of Cytochrome c and Their Regulation in Life and Death Decisions of the Mammalian Cell: From Respiration to Apoptosis. Mitochondrion 2011, 11, 369–381.
- Cui, Z.-G.; Piao, J.-L.; Rehman, M.U.; Ogawa, R.; Li, P.; Zhao, Q.-L.; Kondo, T.; Inadera, H. Molecular Mechanisms of Hyperthermia-Induced Apoptosis Enhanced by Withaferin A. Eur. J. Pharmacol. 2014, 723, 99–107.
- Stankiewicz, A.R.; Livingstone, A.M.; Mohseni, N.; Mosser, D.D. Regulation of Heat-Induced Apoptosis by Mcl-1 Degradation and Its Inhibition by Hsp70. Cell Death Differ. 2009, 16, 638–647.
- Sulpizi, M.; Laio, A.; VandeVondele, J.; Cattaneo, A.; Rothlisberger, U.; Carloni, P. Reaction Mechanism of Caspases: Insights from QM/MM Car–Parrinello Simulations. Proteins Struct. Funct. Bioinform. 2003, 52, 212–224.
- Miscione, G.P.; Calvaresi, M.; Bottoni, A. Computational Evidence for the Catalytic Mechanism of Caspase-7. A DFT Investigation. J. Phys. Chem. B 2010, 114, 4637–4645.
- Hijikata, M.; Kato, N.; Sato, T.; Kagami, Y.; Shimotohno, K. Molecular Cloning and Characterization of a CDNA for a Novel Phorbol-12-Myristate-13-Acetate-Responsive Gene That Is Highly Expressed in an Adult T-Cell Leukemia Cell Line. J. Virol. 1990, 64, 4632–4639.
- Ploner, C.; Kofler, R.; Villunger, A. Noxa: At the Tip of the Balance between Life and Death. Oncogene 2009, 27, S84.
- Oda, E.; Ohki, R.; Murasawa, H.; Nemoto, J.; Shibue, T.; Yamashita, T.; Tokino, T.; Taniguchi, T.; Tanaka, N. Noxa, a BH3-Only Member of the Bcl-2 Family and Candidate Mediator of P53-Induced Apoptosis. Science 2000, 288, 1053–1058.
- Seo, Y.-W.; Shin, J.N.; Ko, K.H.; Cha, J.H.; Park, J.Y.; Lee, B.R.; Yun, C.-W.; Kim, Y.M.; Seol, D.; Kim, D. The Molecular Mechanism of Noxa-Induced Mitochondrial Dysfunction in P53-Mediated Cell Death. J. Biol. Chem. 2003, 278, 48292–48299.
- Wang, Z.; Sun, Y. Identification and Characterization of Two Splicing Variants of Human Noxa. Anticancer Res. 2008, 28, 1667–1674.
- Fei, P.; Bernhard, E.J.; El-Deiry, W.S. Tissue-Specific Induction of P53 Targets in Vivo. Cancer Res. 2002, 62, 7316–7327.
- Saha, M.N.; Jiang, H.; Mukai, A.; Chang, H. RITA Inhibits Multiple Myeloma Cell Growth through Induction of P53-Mediated Caspase-Dependent Apoptosis and Synergistically Enhances Nutlin-Induced Cytotoxic Responses. Mol. Cancer Ther. 2010, 9, 3041–3051.
- Ghavami, S.; Mutawe, M.M.; Sharma, P.; Yeganeh, B.; McNeill, K.D.; Klonisch, T.; Unruh, H.; Kashani, H.H.; Schaafsma, D.; Los, M.; et al. Mevalonate cascade regulation of airway mesenchymal cell autophagy and apoptosis: A dual role for p53. PLoS ONE 2011, 6, e16523.
- Raats, D.A.; de Bruijn, M.T.; Steller, E.J.; Emmink, B.L.; Borel-Rinkes, I.H.; Kranenburg, O. Synergistic Killing of Colorectal Cancer Cells by Oxaliplatin and ABT-737. Cell. Oncol. 2011, 34, 307–313.
- Qin, J.-Z.; Stennett, L.; Bacon, P.; Bodner, B.; Hendrix, M.J.; Seftor, R.E.; Seftor, E.A.; Margaryan, N.V.; Pollock, P.M.; Curtis, A. P53-Independent NOXA Induction Overcomes Apoptotic Resistance of Malignant Melanomas. Mol. Cancer Ther. 2004, 3, 895–902.
- Kim, H.; Rafiuddin-Shah, M.; Tu, H.-C.; Jeffers, J.R.; Zambetti, G.P.; Hsieh, J.J.-D.; Cheng, E.H.-Y. Hierarchical Regulation of Mitochondrion-Dependent Apoptosis by BCL-2 Subfamilies. Nat. Cell Biol. 2006, 8, 1348–1358.
- Flinterman, M.; Guelen, L.; Ezzati-Nik, S.; Killick, R.; Melino, G.; Tominaga, K.; Mymryk, J.S.; Gäken, J.; Tavassoli, M. E1A Activates Transcription of P73 and Noxa to Induce Apoptosis. J. Biol. Chem. 2005, 280, 5945–5959.
- Aikawa, T.; Shinzawa, K.; Tanaka, N.; Tsujimoto, Y. Noxa Is Necessary for Hydrogen Peroxide-Induced Caspase-Dependent Cell Death. FEBS Lett. 2010, 584, 681–688.
- Valis, K.; Prochazka, L.; Boura, E.; Chladova, J.; Obsil, T.; Rohlena, J.; Truksa, J.; Dong, L.-F.; Ralph, S.J.; Neuzil, J. Hippo/Mst1 Stimulates Transcription of the Proapoptotic Mediator NOXA in a FoxO1-Dependent Manner. Cancer Res. 2011, 71, 946–954.
- Alves, N.L.; Derks, I.A.; Berk, E.; Spijker, R.; van Lier, R.A.; Eldering, E. The Noxa/Mcl-1 Axis Regulates Susceptibility to Apoptosis under Glucose Limitation in Dividing T Cells. Immunity 2006, 24, 703–716.
- Lau, L.M.S.; Nugent, J.K.; Zhao, X.; Irwin, M.S. HDM2 Antagonist Nutlin-3 Disrupts P73-HDM2 Binding and Enhances P73 Function. Oncogene 2008, 27, 997–1003.
- Martin, A.G.; Trama, J.; Crighton, D.; Ryan, K.M.; Fearnhead, H.O. Activation of P73 and Induction of Noxa by DNA Damage Requires NF-Kappa B. Aging 2009, 1, 335.
- Santidrián, A.F.; González-Gironès, D.M.; Iglesias-Serret, D.; Coll-Mulet, L.; Cosialls, A.M.; de Frias, M.; Campàs, C.; González-Barca, E.; Alonso, E.; Labi, V. AICAR Induces Apoptosis Independently of AMPK and P53 through Up-Regulation of the BH3-Only Proteins BIM and NOXA in Chronic Lymphocytic Leukemia Cells. Blood 2010, 116, 3023–3032.
- Iglesias-Serret, D.; Piqué, M.; Barragán, M.; Cosialls, A.M.; Santidrián, A.F.; González-Gironès, D.M.; Coll-Mulet, L.; de Frias, M.; Pons, G.; Gil, J. Aspirin Induces Apoptosis in Human Leukemia Cells Independently of NF-ΚB and MAPKs through Alteration of the Mcl-1/Noxa Balance. Apoptosis 2010, 15, 219–229.
- Sheridan, C.; Brumatti, G.; Elgendy, M.; Brunet, M.; Martin, S.J. An ERK-Dependent Pathway to Noxa Expression Regulates Apoptosis by Platinum-Based Chemotherapeutic Drugs. Oncogene 2010, 29, 6428–6441.
- Jin, L.; Tabe, Y.; Kojima, K.; Zhou, Y.; Pittaluga, S.; Konopleva, M.; Miida, T.; Raffeld, M. MDM2 Antagonist Nutlin-3 Enhances Bortezomib-Mediated Mitochondrial Apoptosis in TP53-Mutated Mantle Cell Lymphoma. Cancer Lett. 2010, 299, 161–170.
- DiFeo, A.; Huang, F.; Sangodkar, J.; Terzo, E.A.; Leake, D.; Narla, G.; Martignetti, J.A. KLF6-SV1 Is a Novel Antiapoptotic Protein That Targets the BH3-Only Protein NOXA for Degradation and Whose Inhibition Extends Survival in an Ovarian Cancer Model. Cancer Res. 2009, 69, 4733–4741.
- Chauhan, D.; Tian, Z.; Zhou, B.; Kuhn, D.; Orlowski, R.; Raje, N.; Richardson, P.; Anderson, K.C. In Vitro and in Vivo Selective Antitumor Activity of a Novel Orally Bioavailable Proteasome Inhibitor MLN9708 against Multiple Myeloma Cells. Clin. Cancer Res. 2011, 17, 5311–5321.
- Fernández, Y.; Verhaegen, M.; Miller, T.P.; Rush, J.L.; Steiner, P.; Opipari, A.W.; Lowe, S.W.; Soengas, M.S. Differential Regulation of Noxa in Normal Melanocytes and Melanoma Cells by Proteasome Inhibition: Therapeutic Implications. Cancer Res. 2005, 65, 6294–6304.
- Fribley, A.M.; Evenchik, B.; Zeng, Q.; Park, B.K.; Guan, J.Y.; Zhang, H.; Hale, T.J.; Soengas, M.S.; Kaufman, R.J.; Wang, C.-Y. Proteasome Inhibitor PS-341 Induces Apoptosis in Cisplatin-Resistant Squamous Cell Carcinoma Cells by Induction of Noxa. J. Biol. Chem. 2006, 281, 31440–31447.
- Nikiforov, M.A.; Riblett, M.; Tang, W.-H.; Gratchouck, V.; Zhuang, D.; Fernandez, Y.; Verhaegen, M.; Varambally, S.; Chinnaiyan, A.M.; Jakubowiak, A.J. Tumor Cell-Selective Regulation of NOXA by c-MYC in Response to Proteasome Inhibition. Proc. Natl. Acad. Sci. USA 2007, 104, 19488–19493.
- Núñez-Vázquez, S.; Sánchez-Vera, I.; Saura-Esteller, J.; Cosialls, A.M.; Noisier, A.F.; Albericio, F.; Lavilla, R.; Pons, G.; Iglesias-Serret, D.; Gil, J. NOXA Upregulation by the Prohibitin-Binding Compound Fluorizoline Is Transcriptionally Regulated by Integrated Stress Response-Induced ATF3 and ATF4. FEBS J. 2021, 288, 1271–1285.
- Sosa Seda, I.M.; Mott, J.L.; Akazawa, Y.; Barreyro, F.J.; Bronk, S.F.; Kaufmann, S.H.; Gores, G.J. Noxa Mediates Hepatic Stellate Cell Apoptosis by Proteasome Inhibition. Hepatol. Res. 2010, 40, 701–710.
- Kassis, S.; Grondin, M.; Averill-Bates, D.A. Heat Shock Increases Levels of Reactive Oxygen Species, Autophagy and Apoptosis. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2021, 1868, 118924.
- Hassan, M.; Alaoui, A.; Feyen, O.; Mirmohammadsadegh, A.; Essmann, F.; Tannapfel, A.; Gulbins, E.; Schulze-Osthoff, K.; Hengge, U.R. The BH3-Only Member Noxa Causes Apoptosis in Melanoma Cells by Multiple Pathways. Oncogene 2008, 27, 4557–4568.
- Qin, J.-Z.; Xin, H.; Sitailo, L.A.; Denning, M.F.; Nickoloff, B.J. Enhanced Killing of Melanoma Cells by Simultaneously Targeting Mcl-1 and NOXA. Cancer Res. 2006, 66, 9636–9645.
- Gomez-Bougie, P.; Wuillème-Toumi, S.; Ménoret, E.; Trichet, V.; Robillard, N.; Philippe, M.; Bataille, R.; Amiot, M. Noxa Up-Regulation and Mcl-1 Cleavage Are Associated to Apoptosis Induction by Bortezomib in Multiple Myeloma. Cancer Res. 2007, 67, 5418–5424.
- Holzerland, J.; Fénéant, L.; Banadyga, L.; Hölper, J.E.; Knittler, M.R.; Groseth, A. BH3-Only Sensors Bad, Noxa and Puma Are Key Regulators of Tacaribe Virus-Induced Apoptosis. PLoS Pathog. 2020, 16, e1008948.
- Morales, A.A.; Gutman, D.; Lee, K.P.; Boise, L.H. BH3-Only Proteins Noxa, Bmf, and Bim Are Necessary for Arsenic Trioxide–Induced Cell Death in Myeloma. Blood J. Am. Soc. Hematol. 2008, 111, 5152–5162.
- Yu, J.; Zhang, L.; Hwang, P.M.; Kinzler, K.W.; Vogelstein, B. PUMA Induces the Rapid Apoptosis of Colorectal Cancer Cells. Mol. Cell 2001, 7, 673–682.
- Han, J.; Flemington, C.; Houghton, A.B.; Gu, Z.; Zambetti, G.P.; Lutz, R.J.; Zhu, L.; Chittenden, T. Expression of Bbc3, a pro-Apoptotic BH3-Only Gene, Is Regulated by Diverse Cell Death and Survival Signals. Proc. Natl. Acad. Sci. USA 2001, 98, 11318–11323.
- Fricker, M.; O’prey, J.; Tolkovsky, A.M.; Ryan, K.M. Phosphorylation of Puma Modulates Its Apoptotic Function by Regulating Protein Stability. Cell Death Dis. 2010, 1, e59.
- Yu, J.; Zhang, L. PUMA, a Potent Killer with or without P53. Oncogene 2008, 27, S71–S83.
- Jeffers, J.R.; Parganas, E.; Lee, Y.; Yang, C.; Wang, J.; Brennan, J.; MacLean, K.H.; Han, J.; Chittenden, T.; Ihle, J.N. Puma Is an Essential Mediator of P53-Dependent and-Independent Apoptotic Pathways. Cancer Cell 2003, 4, 321–328.
- Happo, L.; Cragg, M.S.; Phipson, B.; Haga, J.M.; Jansen, E.S.; Herold, M.J.; Dewson, G.; Michalak, E.M.; Vandenberg, C.J.; Smyth, G.K. Maximal Killing of Lymphoma Cells by DNA Damage–Inducing Therapy Requires Not Only the P53 Targets Puma and Noxa, but Also Bim. Blood J. Am. Soc. Hematol. 2010, 116, 5256–5267.
- Mintoo, M.; Khan, S.; Wani, A.; Malik, S.; Bhurta, D.; Bharate, S.; Malik, F.; Mondhe, D. A Rohitukine Derivative IIIM-290 Induces P53 Dependent Mitochondrial Apoptosis in Acute Lymphoblastic Leukemia Cells. Mol. Carcinog. 2021, 60, 671–683.
- Muller, P.A.; Vousden, K.H. P53 Mutations in Cancer. Nat. Cell Biol. 2013, 15, 2–8.
- Charvet, C.; Wissler, M.; Brauns-Schubert, P.; Wang, S.-J.; Tang, Y.; Sigloch, F.C.; Mellert, H.; Brandenburg, M.; Lindner, S.E.; Breit, B. Phosphorylation of Tip60 by GSK-3 Determines the Induction of PUMA and Apoptosis by P53. Mol. Cell 2011, 42, 584–596.
- Wu, B.; Qiu, W.; Wang, P.; Yu, H.; Cheng, T.; Zambetti, G.P.; Zhang, L.; Yu, J. P53 Independent Induction of PUMA Mediates Intestinal Apoptosis in Response to Ischaemia–Reperfusion. Gut 2007, 56, 645–654.
- Michalak, E.M.; Villunger, A.; Adams, J.M.; Strasser, A. In Several Cell Types Tumour Suppressor P53 Induces Apoptosis Largely via Puma but Noxa Can Contribute. Cell Death Differ. 2008, 15, 1019–1029.
- Chen, S.; Lóssio, C.F.; Verbeke, I.; Verduijn, J.; Parakhonskiy, B.; Van der Meeren, L.; Chen, P.; De Zaeytijd, J.; Skirtach, A.G.; Van Damme, E.J. The Type-1 Ribosome-Inactivating Protein OsRIP1 Triggers Caspase-Independent Apoptotic-like Death in HeLa Cells. Food Chem. Toxicol. 2021, 157, 112590.
- Wei, J.; O’Brien, D.; Vilgelm, A.; Piazuelo, M.B.; Correa, P.; Washington, M.K.; El-Rifai, W.; Peek, R.M.; Zaika, A. Interaction of Helicobacter Pylori with Gastric Epithelial Cells Is Mediated by the P53 Protein Family. Gastroenterology 2008, 134, 1412–1423.
- Fischer, S.F.; Belz, G.T.; Strasser, A. BH3-Only Protein Puma Contributes to Death of Antigen-Specific T Cells during Shutdown of an Immune Response to Acute Viral Infection. Proc. Natl. Acad. Sci. USA 2008, 105, 3035–3040.