Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 3 by Camila Xu and Version 4 by Camila Xu.
Phenolic metabolites are organic compounds with at least one or more hydroxyl groups attached to arylic systems with simple variations to highly polymerized molecules.
- marine natural products
- phenolics
- anticancer
1. Phenolic Compounds of Marine Origin—General Characteristics and Biosynthesis
Phenolic metabolites are organic compounds with at least one or more hydroxyl groups attached to arylic systems with simple variations to highly polymerized molecules [1][2]. They are often found in terrestrial plants as well as in red, brown and green macroalgae (seaweeds) [1], sponges [3][4], corals [5][6][7][8] and microalgae [9]. The recent studies have shown that marine-derived microorganisms also produce these phytochemicals [10], what could be confirmed by a metagenomic analyses [11]. In addition to their primary role in plant physiology for structural scaffolding and maintenance of the plant integrity as well as in the development of the plant through lignin and pigment biosynthesis, the phenolic compounds have an important role as secondary metabolites in protection against stress, defence against predators, repellent or extermination of microorganisms or other pathogens, the fouling process and UV radiation [1]. Phenols’ structural versatility is the result of modifications of the basic phenolic structure obtained through glycosylation, oxygenation, hydroxylation, methylation, unsaturation, isomerization, and oligomerization or polymerization [12], as well as the variations in the oxidation state of the pyran ring as observed for flavonoids [13]. Many of the natural phenols are glycosylated while the toxic effect is exerted by the aglycone part [14]. Due to unique structures and diverse scaffolds containing halogen atoms and the possibility of polymerization into multiple forms, all contributing to broad pharmacological activities, phenolic compounds of marine origin have attracted the great attention of the scientific community [1].
The most recent and comprehensive review by Mateos and colleagues presented an insight into marine phenolics regarding their antidiabetic, antioxidant, antiviral, antimicrobial, anti-inflammatory and anticancer activities [15]. However, their activities are largely affected by pharmacokinetic and pharmacodynamic properties, especially bioavailability, absorption and metabolic enzymatic interactions. Some of those are observed only in in vivo studies, since in vitro assays might result in inadequate results and conclusions [13]. Poor bioavailability of phenols is mostly due to rapid and extensive metabolism and rather poor passage through the blood brain barrier or, in case of quercetin, the low absorption of the bioactive aglycone part in the gastrointestinal tract [16]. Moreover, certain phenolic acids like caffeic or syringic acid, as well as coumarin derivatives, were predicted by in silico methods to be more hydrophobic, thus capable to pass the brain barrier to a greater extent [17].
Regardless of the therapeutic potential of this class of compounds, it should be noted that phenolics from marine species are significantly less explored than their terrestrial counterparts. Their isolation is often challenging because of the instability and the high chemical reactivity, and often, the additional necessity for the employment of new technologies for the isolation as well as the establishment of an appropriate analytical identification and characterization method [2]. In a recent review, Getachew and co-workers compared novel and green extraction processes with the conventional techniques that are usually time consuming and use the large amounts of organic solvents. The authors showed that the employment of the extraction procedures assisted by ultrasound, microwave or enzyme as well as pressurized liquid and supercritical fluid (ScCO2) extractions gave phenolics in higher isolated yields in a shorter time and with a smaller consumption of solvents compared to the classical extraction methods [18].
Organism collection and supply, together with low yields and small amounts of isolated natural products, are the main challenges in marine bioprospecting. The development of the efficient syntheses of the desired biologically active metabolites, with the aim of preserving the marine ecosystem, is thus often essential [19]. The understanding of secondary metabolites biosynthesis in organisms could aid to the establishment of bioinspired synthesis and development of new synthetic strategies [20].
Phenols in nature can be synthesized through three known pathways, connecting the metabolism of primary carbohydrates and amino acids with secondary metabolites: the shikimic acid-phenylpropanoid pathway, the malonate-acetate (polyketide) pathway and mevalonate-acetate (isoprenoid) pathway (Scheme 1) [21][22]. The malonic acid pathway is significantly expressed in microorganisms, both, fungi and bacteria [23]. The shikimic acid-phenylpropanoid pathway is the major biosynthetic route for the formation of most of phenols, especially in vascular plants.
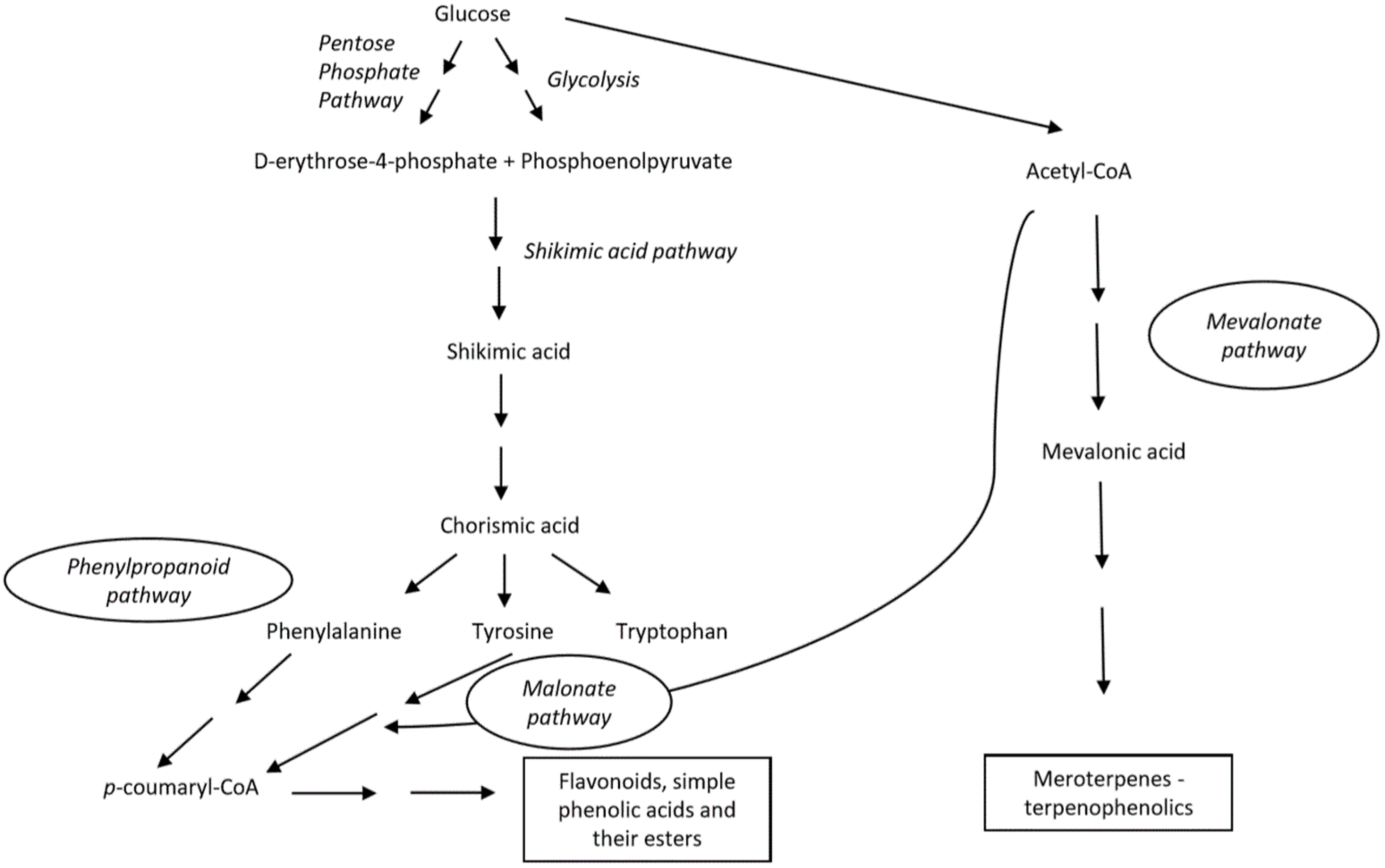
Scheme 1. Main biosynthetic pathways for generation of phenolic compounds.
The shikimate pathway initially involves the synthesis of aromatic amino acids (phenylalanine, tyrosine and tryptophan) via the modification of chorismic acid, obtained from carbohydrate precursors in glycolysis. The biosynthesis of phenylpropanoid partly involves distinct enzymes that might be differently expressed depending on both, biotic and abiotic features, and thereby can induce additional complexity to the produced compounds. The key enzyme is phenylalanine ammonia-lyase (PAL), which is responsible for the formation of cinnamic acid. Next, cinnamic acid is transformed into p-coumarid acid by cinnamate 4-hydroxlyase (C4H) [20][23][24].
Similarly to previous pathways, in the malonic acid pathway, the tyrosine is deaminated to p-coumaric acid by the key enzyme, tyrosine ammonia lyase (TAL). In addition, p-coumaric acid is functionalized by CoA to produce p-coumaroyl-CoA by 4-coumaroyl CoA ligase (4CL) and further reacts with three molecules of malonyl-CoA obtained from acetyl-CoA to give chalcone by chalcone synthase. The obtained tetraoxychalcone is transformed to flavanone–naringenin that serves as a precursor of many other flavonoids [25][26]. By both of these pathways, flavonoids, simple phenolic acids and their esters are being produced.
Kumari et al. summarized the mevalonate-acetate isporenoid pathway in which basic isoprene (C5) units are formed [27]. The latter can be also obtained via a non-mevalonate route. Further condensation of such units in linear or branched intermediates that, eventually, undergo multi-step cyclization reactions catalyzed by terpene synthases and generate miscellaneous terpenoids. As authors elaborated, such a biosynthetic pathway leads to the formation of isoprene units as fundamental structural elements of terpenoids, but also phenols and alcohols [27].
The biosynthesis of natural phenolics in terrestrial plants has been more studied and better understood compared to biogenesis of phenols present in marine organisms [2]. In recent decades a significant progress has been made regarding that issue [28][29][30]. The role of vanadium haloperoxidases that are responsible for the incorporation of bromine in marine natural products was highlighted. The reaction occurs in two steps, including the generation of enzymatically formed BrOH which then promotes an electrophilic attack (bromination) of certain organic molecules [28]. Another interesting group of marine phenolic compounds, phlorotannins are formed by the polymerization of 1,3,5-trihydroxybenezene units (phloroglucinol, 1) into more complex structures [29]. However, the complete mechanism is poorly understood and remains to be discovered, despite Meslet-Cladière et al. proposing the key step in phlorotannin biosynthesis by analyzing the brown alga, Ectocarpus siliculosus. As demonstrated by the authors, a polyketide synthase (type III, PKS) is essential for the synthesis of the phloroglucinol unit from malonyl-CoA of the malonic acid pathway. Further, they observed a positive correlation between the PKS gene and the quantity of the phloroglucinol compound [30].
2. Anticancer Properties of Marine-Derived Phenolic Compounds
Marine-derived phenolics exhibit a wide range of biological effects as a consequence of structural uniqueness and complexity [15]. According to Jimenez-Lopez et al., those effects arise mostly from their antioxidant activity, in particular, the ability to scavenge reactive oxygen species (ROS) by a direct H-atom donation process or single electron transfer (SET) from the hydroxyl groups followed by protonation [31]. The latter has been extensively computationally studied regarding the thermodynamic and kinetic properties of the transfer reaction by Di Meo and co-workers [32]. Furthermore, since redox homeostasis is impaired in tumor tissue, which at a certain (moderate) concentration can initiate the growth and progression of cancer cells, ROS represents a potential target for chemotherapeutics [33]. On the other side, anticancer activity of marine phenols could be achieved by the suppression of the telomerase expression and activity or by the modulation of the proliferation signal pathways as reported and reviewed by others [15][34]. Numerous research studies have been conducted so far, investigating the anticancer activity of organic or water extracts obtained mostly from macro- and microalgae, sponges and tunicates [35][36][37][38][39][40][41][42][43]. As admonished by Rosa et al., it is necessary to characterize the chemical composition so that the activity can be associated with the presence of a certain metabolite. The same authors have pointed out a scarce number of in vivo studies and clinical trials of phenolic metabolites to date [44]. As the backbone in the development of new drugs, this research aims to provide an overview of the recently isolated and characterized marine phenolic compounds together with their most significant antitumor effects. Phenolic metabolites are divided into seven classes as following: phlorotannins, bromophenols, flavonoids, coumarins, terpenophenolics, quinones and hydroquinones and miscellaneous compounds. Importantly, the nomenclature of compounds, more precisely the numbering of the positions of atoms and substituents, is retained as in the original research papers. Some of the compounds can be classified into several different groups, confirming a complex division of natural-derived products.2.1. Phlorotannins
Phlorotannins 1–7 are a class of phenolic compounds similar to terrestrial-derived tannins, recognizable by the high proportion of hydroxyl groups with elevated water solubility and a binding affinity for the biological macromolecules such as proteins and polysaccharides (Figure 1) [44][45][46][47]. They are biosynthesized by marine brown macroalgae (Phaeophyta) and can be considered as primary and secondary metabolites, with the former resulting from their role in the formation of the cell wall based on the phlorotannin-alginic acid complex. Their secondary metabolite functions include protection from infections and ultraviolet radiation by high absorbance of the UV-B spectrum, as well as defense against herbivores [45]. Phlorotannins are formed by the polymerization ofna monomer unit, phloroglucinol (1), thereby achieving a wider mass range when compared to the terrestrial counterparts. The monomers can combine through carbon-oxygen or carbon-carbon bonds at different positions and to different degrees, forming linear, branched and cyclic compounds [45]. Such structures of phlorotannins are highly susceptible to pH, light, temperature and oxidation, causing an urgent need to develop new technologies in the field of nano science and green chemistry strategies [48]. Therefore, a novel method of encapsulation has been investigated using polyvinylpyrrolidone nanoparticeles, which resulted in enhanced functions of phlorotannins with regards to a reduced kinetic release and toxicity towards normal cells and greater antiradical activity [49]. Just recently, Kaushalya and Gunathilake have isolated phlorotannins from the brown algae, Sargassum ilicifolium, and performed encapsulation in the chitosan-tripolyphospahte carrier. Those particles were found to exhibit higher antioxidant activities and total phlorotannin content upon in vitro digestion, indicating the potential for the targeted delivery of phlorotannins [50].
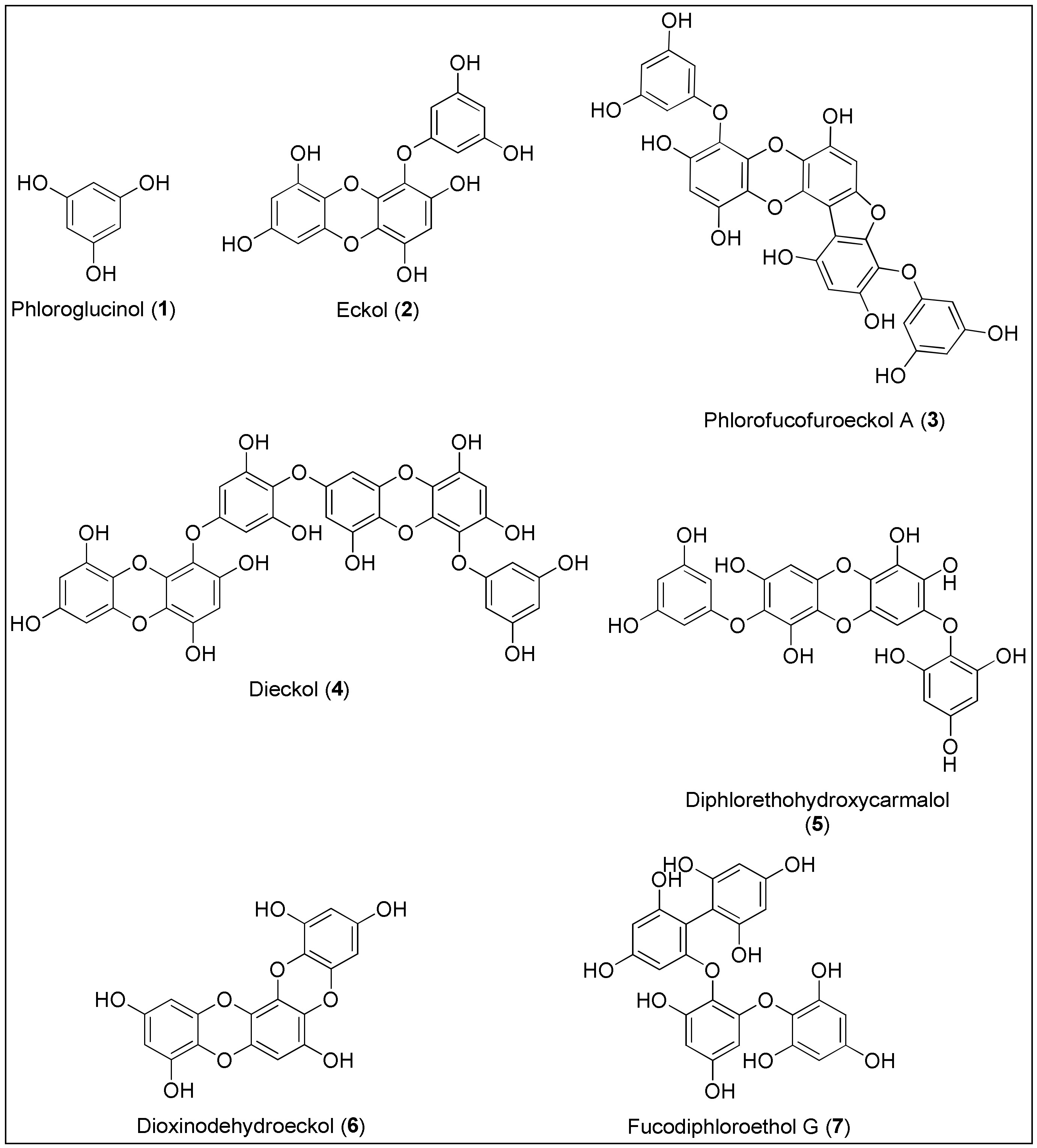
Figure 1. Chemical structures of phlorotannins 1–7 with anticancer activities isolated from brown seaweeds.
Phlorotannins are classified into four main groups based on the linkage functionality between aromatic units. Fuhalols and phlorethols possess an ethereal linkage, fucols aromatic, fucophlorethols ethereal and aromatic linkages, whereas eckols and carmalols are bridged through a 1,4-dioxin linkage [47]. Due to biological potential and the possibility of the atropoisomeric chirality because of the restricted rotation, these molecules have attracted considerable attention of synthetic chemists [51]. As stated by Erpel and her team in a comprehensive review on phlorotannins’ isolation and therapeutic potential, this class of phenolics exerts anticancer activity by various biological mechanisms, leading to apoptosis and inhibition of angiogenesis and tumor metastasis [46].
Phloroglucinol (1) was primarily isolated from marine brown alga, Ecklonia cava (Kjellman, 1885), but it is also produced by other species belonging to the classes of Phaeophyceae and Fucaceae [52][53]. Phloroglucinol (1), displayed antioxidant, antibacterial, enzyme inhibition and anticancer activities [45]. In 2012, Kwon and colleagues were the first who conducted a study on phloroglucinol’s potential to inhibit tumor angiogenesis in vitro and in vivo. As reported, phenol 1 inhibited the migration and capillary-like tube formation of endothelial progenitor cells’ processes depending on the vascular endothelial growth factor (VEGF) which resulted in the reduction of Lewis lung carcinoma in a mouse model. To obtain more information regarding cancer growth and metastatic spread, the additional evaluation of the mechanism that triggers the inhibition has to be conducted [54]. Although phenol 1 displayed 50% inhibition on the invasive and migratory ability of the MDA-MB-231 breast cancer cells at a concentration of 50 µM, Kim et al. reported that it did not initiate their significant death. The authors also demonstrated that treatment with 1 led to the downregulation of SLUG protein in the aforementioned MDA-MB-231 and BT549 breast cancer cell lines, via PI3K⁄Akt and Ras⁄Raf-1⁄ERK signalling pathways. As suggested, SLUG is responsible for the regulation of epithelial-mesenchymal cell transition, and thus for invasiveness of cancer cells [55]. The Kang’s group conducted two studies in 2014, invetigating the pro-apoptotic activity of phenol 1 against HT-29 colon cancer cells [56][57]. They observed that apoptosis of the cells occurred through an insulin-like growth factor 1 receptor (IGF-1R) and the inhibition of downstream proteins, similarly to the results of Kim et al. [57]. Phenol 1 also altered the expression of proteins belonging to the Bcl-2 family; more precisely, it induced the upregulation of pro-the apoptotic proteins Bax and Bad and the downregulation of anti-apoptotic Bcl-2 and Bcl-xL [56]. In 2015, Kim and co-workers reported that 1 exhibited anticancer activity against breast cancer stem-like cells by reducing the expression of CD44, Oct4, Notch2, β-catenin and Sox2 proteins. The latter is often connected to the development of resistance against chemotherapeutics. The treatment with phenol 1 enhanced the sensitivity of cells to the ionizing radiation and standard chemotherapeutic drugs (taxol, cisplatin and etoposide) [58]. The higher sensitization of colon cancer cells, HT-29 and HCT116, to 5-fluorouracil (5-FU) was also observed by Lopes-Costa et al. after treatment with 1. Interestingly, the authors also noticed that phloroglucinol (1) decreased apoptosis of cells at a lower concentration which can be explained by the scavenging activity of phenol against ROS, induced by 5-FU [59].
Another phlorotannin isolated from Ecklonia cava, eckol (2), was found to exhibit radioprotective properties in vitro by ROS scavenging [60] and in vivo through the reduction of side effects caused by gamma ray-irradiation of entire mouse bodies, more precisely, the repair of damaged DNA and the recovery of hematopoiesis [61]. Based on these results, Park’s group suggested that eckol (2) could be used as an adjuvant therapy to mitigate the side effects of radiation in cancer patients [60][61]. In 2011, Hyun et al. assayed 2 for targeting stem-like glioma cells. Eckol (2) decreased the expression of CD133, Nestin and Musashi-1, which represent markers of stem cells. Further, treatment with 2 increased the sensitivity of glioma cells to the cytotoxic prodrug, temozolomide, as well as to ionizing radiation via PI3K⁄Akt and Ras⁄Raf-1⁄ERK signalling pathways, similarly to phloroglucinol (1) (vide supra) [62]. Moreover, phlorotannin 2 significantly inhibited the growth of the pancreatic cancer cell line (SW1990) induced by the presence of an exogenous regenerating gene protein (Reg3A). The biological importance of the Reg3A protein is evident in the influence on pancreatic cancer cells’ proliferation and progression, additionally with the appearance of an inflammatory microenvironment [63]. Furthermore, another member of pancreatitis-associated proteins, Reg3g, was upregulated in an experimental colitis mouse model when treated with eckol (2) at doses of 0.5–1.0 mg/kg. Since Reg3g appeared to be a protection against the colon injury development, the authors suggested the possibility of using 2 as an alternative treatment of colitis [64]. A relation between the pharmacological effects of eckol (2) and the modulation of the innate and adaptive immune systems was also observed by Zhang and co-workers in vivo on a mouse model bearing a sarcoma (S180) xenograft. Namely, a phlorotannin 2 administration resulted in the increase of cytotoxic T-lymphocytes, the induction of a type 1 helper T cell (Th1), which is required for the annihilation of the tumor and the activation of dendritic and mononuclear phagocytic cells [65]. Finally, Cho et al. very recently demonstrated protective activity of eckol (2), in vitro, against skin inflammation promoted by a mixture of TNF-α and IFN-γ, further emphasizing the importance of 2 in inflammatory processes and responses [66].
In 2012, Eo et al. performed a mechanistic study regarding the induction of apoptosis in human colorectal cancer cell lines with the derivative of eckol (2), phlorofucofuroeckol A (3) [67]. The results showed that at 100 μM, there were 3 decreased cells viabilities of 38%, 31%, 47% and 90%, for HCT116, HT-29, LoVo and SW480 cancer cells, respectively, with the SW480 cells being the most sensitive to 3. Furthermore, the authors assumed that cell viability might have occurred via apoptosis since its biomarker, cleaved poly (ADP-ribose) polymerase (PARP) was increased. Furthermore, treatment with 3 led to the enhanced expression of a possible therapeutic target, activating transcription factor 3 (ATF3) [67]. Lee’s group extracted phlorofucofuroeckol A (3) from E. cava and observed the suppression of migration and invasion of MCF-7 and MDA-MB-231 breast cancer cell lines by the downregulation of Nf-κB activity and Toll-like receptor 4 (TLR-4) signalling. The latter is shown to be crucial in the development of breast cancer. Moreover, the results indicated that 3 reduced the expression of matrix metalloproteinases (MMPs) 2 and 9, also involved in invasion and metastasis processes [68]. The most recent study carried out by Manandhar et al., investigated the activity of 3, isolated from Ecklonia stolonifera, against a mouse melanoma cell line B16F10. However, phlorofucofuroeckol A (3) did not exhibit a significant antiproliferative effect towards the above mentioned cancer cells [69].
Between all phlorotannins, the anticancer properties of dieckol (4) have been the most comprehensively evaluated. Similarly to phlorofucofuroeckol A (3), dieckol (4) also induced a reduction in invasive potential of MCF-7 cells [68]. Zhang and colleagues studied the cytotoxicity of 4, the effect on invasiveness of human fibrosarcoma cells (HT1080) as well as its influence on the expression of MMP-2 and -9. The treatment of HT1080 cells with dieckol (4) did not result in a general toxic outcome even at the highest concentration of 250 µM. Furthermore, 4 also downregulated the expression of the tested matrix metalloproteinases through Nf-κB signalling [70]. Interestingly, analogous observations were made by Oh and co-workers during the examination of the inhibitory effect of dieckol (4) against hepatocellular carcinoma cells (SK-Hep1). The authors also reported that the negative effect of 4 on MMP-9 could have arisen from the modulation of the MAPK signalling pathways [71]. Importantly, very recently all molecular mechanisms underlying MMP inhibition (MAPK, activator protein-1 and NF-κB signalling) were confirmed by Wang’s group by using a human dermal fibroblast cell line (HDF) [72]. In 2012, Park et al. performed two studies and analyzed a new anticancer strategy by employing dieckol (4), regarding the inhibition of ROS and migration potential of the malignant and non-metastatic melanoma cells, B16F10 and B16F0, respectively [73][74]. The authors observed that the higher concentration of ROS (in particular, H2O2) enhanced the migration of B16F0, which was in relation to increased levels of the Ras-related C3 botulinum toxin substrate 1 (Rac1) and the Wiskott–Aldrich syndrome protein family member 2 (WAVE2). Contrary, treatment with 4 diminished those effects accompanied by the inhibition of actin polymerization [73]. Both proteins are included in the signalling pathway associated with actin reorganization and cell motility and, consequently, the metastasis of cancer cells [75]. The latter is in agreement with the results from the second study where 4 suppressed the migration of HT1080 cells via reduced expression and phosphorylation of focal adhesion kinase (FAK) which is also related to the migration potential of the cell [74]. Dieckol (4) can also cause the selective anticancer activity against hepatocellular carcinoma cell lines (Hep3B, Sk-Hep1). At a concentration of 100 µM, 4 induced apoptosis of 62.2% of Hep3B cells. Furthermore, Yoon et al. proposed signalling pathways, underlying apoptosis, which are manifested in the activation of mitochondrial-related release of cytochrome c and death receptor-mediated caspase cascade as revealed by immunoblotting analyses [76]. In 2015, an interesting study was conducted by Li and co-workers in which dieckol (4) acted as antiproliferative and antiangiogenic agent through the inhibition of the mentioned MMP-2 and -9, MAPK signalling proteins, ERK, and p38, additionally targeting VEGF. The authors used non-transformed, human umbilical vein endothelial cells (EA.hy926), of which the viability was not significantly decreased by 4. In vitro results were compared to computational docking calculations which showed that 4 accomplishes binding to aforementioned proteins via predominantly formed hydrogen bonds with certain amino acids [77].
The antitumor activity of the dieckol (4), resulting from the influence of this phlorotannin on proteins involved in the apoptotic signalling pathway and from the induced oxidative stress, was also studied by Ahn’s team [78]. They reported the IC50 values of 77.3 and 92.7 µM in ovarian cancer cells, A2780 and SKOV3, respectively. Therewithal, the antitumor efficacy was also evaluated in vivo using mice bearing ovarian tumors, the volume of which was significantly decreased after a four-week treatment with 4, without inducing side effects in terms of liver and kidney toxicity [78]. Moreover, in 2015, Kim’s group applied the centrifugal partition chromatography to isolate three members of eckol subclass of phlorotannins from E. cava. Among them, dieckol (4) exhibited the highest anticancer activity against human breast cancer cell line, MCF-7 cells, wherein the IC50 value was between the two highest concentrations (64 and 128 µM). The gap-closure assay demonstrated that the mobility of the cells was significantly lower upon treatment with 4. That observation was corroborated by the increased expression of glycoproteins, TIMP-1 and -2 which are inhibitors of MMP and involved in the degradation of extracellular matrix (ECM), thus contributing to the suppression of cell migration. These results also matched with the downregulation of MMP-9 [79].
You et al. used the cell proliferation reagent (WST-1) to examine the dependence of viability of MCF-7 and SK-BR-3 breast cancer cells on various concentrations of dieckol (4). Contrary to the Kim’s report from 2015 mentioned before, 50% of cell death was reached at concentration values higher than 200 µM. An increase in Bax/Bcl-2 ratio indicating mitochondrial-mediated apoptosis was observed, but only in SK-BR-3 cells. Thus, further studies have to be undertaken [80]. Phlorotannin 4 was found to exert protective and chemopreventing effects against hepatocellular carcinoma (HCC) in rats induced by N-nitrosodiethylamine (NDEA) administration, as reported by Sadeeshkumar et al. [81][82]. At a dosage of 40 mg/kg, 4 prevented lipid peroxidation, increased antioxidant activity and reversed the activities of certain hepatic marker enzymes which the authors hypothesized as a consequence of scavenging capacity of dieckol (4) [82]. The same group analyzed the molecular mechanisms that mediated observed NDEA-induced hepatocarcinogenesis in male Wistar rats. Phlorotannin 4 also displayed potential chemoprevention through the modulation of angiogenesis, invasion, apoptosis and inflammation via the upregulation of VEGF, MMP-2/9, proliferating cell nuclear antigen (PCNA) and COX-2 (cyclooxigenase-2), respectively [81]. In addition to breast, melanoma and hepatocellular cancer cells, dieckol (4) was also screened for anticancer properties against non-small-cell lung cancer (A549) [83] and human pancreatic cancer cells (PANC-1) [84] in the two most recent studies. At concentrations of 25 µg/mL and 20 µM, 4 induced 50% of A549 and PANC-1 cells deaths, respectively [83][84]. Wang and colleagues demonstrated the anti-migratory and apoptotic activity of 4 in A549 cells, associated with the inhibition of the Pi3K/AKT/mTOR signalling pathway and activation of the tumor-suppressor, E-cadherin [83]. Lastly, Xu and co-workers proved that 4 could be used to treat pancreatic cancer since it increased the expression of proapoptotic protein (Bax) and decreased antiapoptotic Bcl2 protein as well as cyclin D1, whose overexpression is often related to chemoresistance [84]. Finally, deickol (4) was found to provide the protection from gamma radiation and consequent damage, both in vitro and in vivo [85][86].
In 2004, Toume et al. isolated and characterized diphlorethohydroxycarmalol (5) from another brown alga, Ishige okamurai. Carmalol derivative 5 displayed moderate cytotoxic activity towards vincristine-resistant as well as sensitive murine leukemia cells (P-388) with reported IC50 values being 8.0 and 10.5 µg/mL [87]. Yeon’s group was the first to assess the anticancer activity of 5 against promyelocytic leukemia cells (HL60) and study its apoptotic mechanism. According to the authors, the compound 5 induced concentration-dependent growth inhibition with the IC50 value lower than 25 µg/mL. Western blot analysis demonstrated that the treatment of HL60 cells with 5 resulted in an increased expression of cleaved caspase-3 and cleaved PARP as biomarkers for cell apoptosis. Furthermore, the upregulation of pro-apoptotic Bax and downregulation of anti-apoptotic Bcl-2 were also observed [88]. Similarly, these results were also confirmed by Park et al. by using the mouse embryonic fibroblast cell line (3T3-L1) [89]. Interestingly, several studies indicated photoprotective activity of diphlorethohydroxycarmalol (5) against UVB radiation and side effects, through the modulation of antioxidant system, absorption of radiation, inhibition of MMPs expression and scavenging of ROS, thus, demonstrating a wider possibility of biological action of 5 [90][91][92][93].
Another component of the Ecklonia species, an eckol derivative dioxinodehydroeckol (6), exhibited significant antiproliferative activity against MCF-7 with regard to MDA-MB-231 cells with 50% inhibition of proliferation induced at 5–10 and 50–100 µM, respectively. As studied by Kong et al., 6 induced apoptosis in MCF-7 cells via the regulation of the NF-κB pathway which eventually caused the downregulation of p65, IKK (IκB kinase) and NIK (NF-κB inducing kinase). The apoptotic activity of 6 was also associated with increased activities of caspase-3 and -9 and enhanced cleavage of a caspase substrate, PARP. The treatment with 6 induced a higher expression of pro-apoptotic Bax and lower expression of anti-apoptotic protein, Bcl-2 [94]. Contrary to these results and at the same time, Yoon and co-workers observed that the dioxinodehydroeckol (6) was neither effective on melanoma cells (B16F10) nor it affected melanin biosynthesis in melanoma cells [95]. Interestingly, Lee and colleagues came to opposite conclusions when screening for anti-melanogenic activity of 6 in B16F10 cells. According to the authors, the phlorotannin 6 inhibited melanin production and tyrosinase activity with reported IC50 values of 19.3 ± 1.2 and 24.2 ± 0.9 µM, respectively. Western blot and RT-PCR analyses revealed that the compound 6 downregulated tyrosinase and tyrosinase-linked proteins (TRP-1 and TRP-2) as well as MITF (microphthalmia-associated transcription factor), which are all involved in the regulation of melanin formation. Nevertheless, the viability of melanoma cells was not significantly changed by 6 [96]. The same modulation of apoptotic proteins and of the Bcl-2 family together with a caspase-dependent pathway was demonstrated by Ryu et al. in human keratinocyte cells (HaCaT) which were exposed to UV-B radiation. Based on those results, dioxinodehydroeckol (6) could be considered as a protective compound against radiation-induced skin damage [97].
Ham and colleagues were the first to isolate a phlorotannin metabolite with phenyl and ether bridges connecting phloroglucinol units, named fucodiphloroethol G (7), from a methanol extract of E. cava [98]. A comprehensive screening of antiproliferative properties of 7 against human cell lines was performed by Li’s group who noticed a specific activity towards cancer cells rather than a normal fibroblast cell line (MRC-5). At concentration values of 298.2, 226.5, 242.5 and 228.5 µM, fucodiphloroethol G (7) caused 50% inhibition of HeLa (human cervical cancer), A549, HT1080 and HT29 (colon carcinoma) cancer cells, respectively [99]. This was preceded by another research performed by Li et al., when assaying 7 against leukemia cells (HL-60). However, they used a much lower concentration (≤100 µM) and concluded that treatment with polyphenol 7 did not cause any significant cytotoxic effects against the aforementioned cell line [100]. Mostly the same research group conducted an extensive study of the molecular basis of angiogenesis suppression induced by 7 in human umbilical vein endothelial cells (EA.hy926 and ECV-304), in which angiogenic activity was stimulated by exogenous VEGF. Results have demonstrated that fucodiphloroethol G (7) blocked the MAPK and Akt signalling pathway necessary for the expression of proteinases (MMP-2, MMP-9 and APN) involved in the degradation of the extracellular matrix. Since the latter is crucial for invasion and cancer spreading, this signalling protein represents novel targets for therapies of metastatic cancers [101].
2.2. Bromophenols
More than 5000 halogenated natural products have been isolated and characterized, most of which are biosynthesized by various marine organisms (algae, corals, sponges and marine-associated microorganisms) [102]. The presence of halogen atom substituents often leads to better biological activities due to the possibility of the formation of various interactions between target and ligand, which include hydrogen and halogen bonds as well as other polar–polar interactions. The presence of halogen atoms may also cause conformational changes in molecules, the enhancement of lipophilicity and an increased affinity and binding constant of ligand to target, thus attracting synthetic and medicinal chemists to study and to develop functional foods as well as to design new drugs [103][104]. More than 600 halogenated compounds of a marine origin have been presented in a comprehensive review by Gordon W. Gribble, summarizing their antiparasitic, antiviral, anti-inflammatory, antibacterial, anticancer and antioxidant activities [102]. One of the interesting class of organohalogens are bromophenols 8–29 (Figure 2). These compounds can be characterized by the diverse phenolic skeletons bearing at least single bromine substituent [105][106]. Even though bromophenolics have been isolated from green, brown and red algae, the latter have been found to contain brominated compounds as one of the major class of metabolites [28].
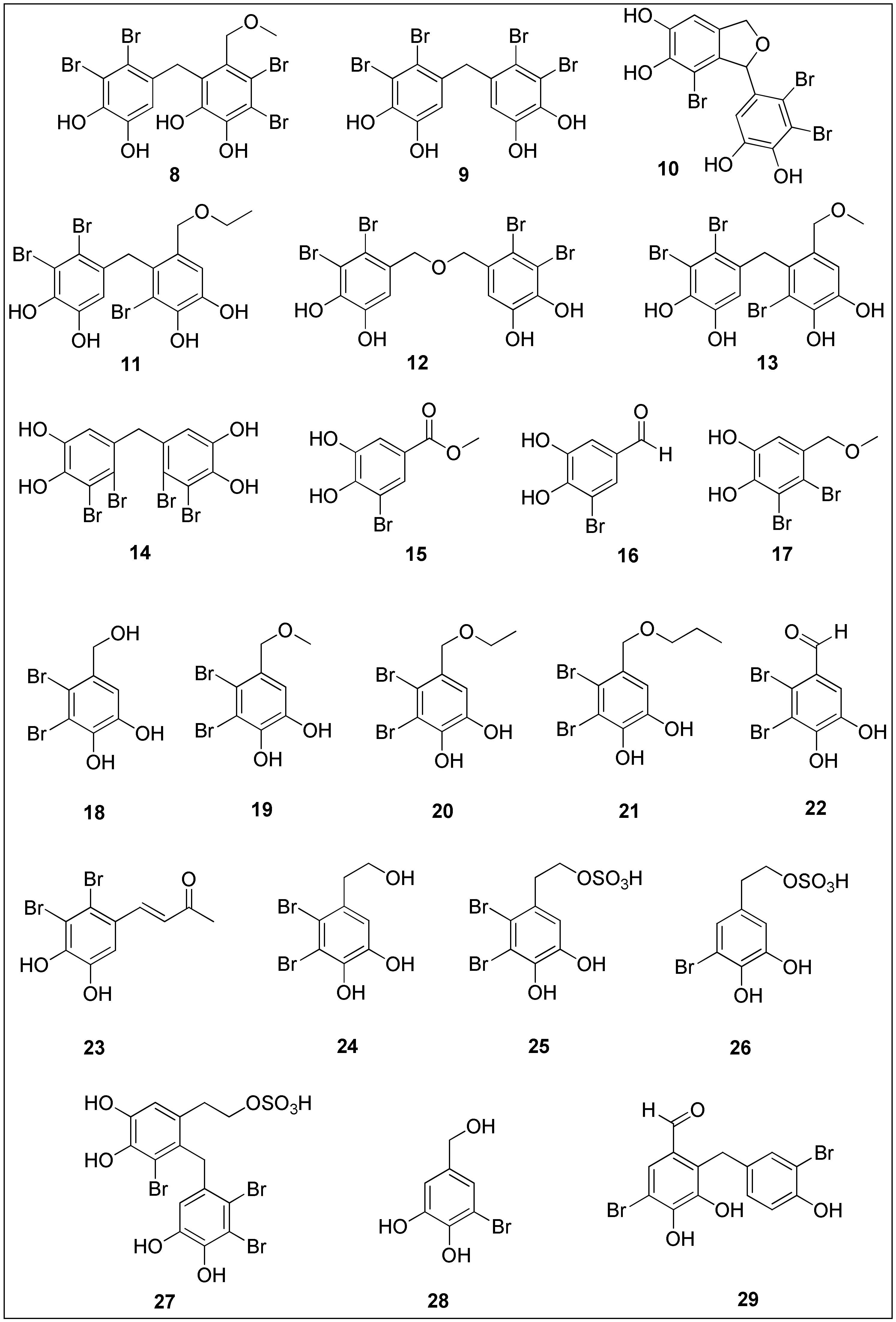
Figure 2.
Chemical structures of bromophenols
8
–
29
with significant anticancer activities isolated from brown, red and green seaweeds.
Bromination can possibly lead to the improvement of bioselectivity [107]. For example, in 2004, Xu and co-workers [108] isolated several dibenzyl bromophenols 8–13 from an ethanolic extract of marine brown alga, Leathesia nana. These bromophenols exhibited moderate antiproliferative activity against several cancer cell lines with IC50 values in a nanomolar range, notwithstanding the fact that no activity of preliminary screened ethanolic extract was observed. At concentrations of 1.8, 3.8, 2.7 and 2.2 nM, bromophenol 9 inhibited 50% of A549, BGC-823, MCF-7 and HCT-8, respectively. Moreover, treatment of hepatoma cells (Bel7402) with compounds 8, 11 and 12 resulted in IC50 values between 4.8 and 7.4 nM [108]. The same group conducted a broader study in 2009, evaluating the cytotoxicity of compounds 8–13 against human malignant melanoma (B16-BL6), human sarcoma (HT-1080) and A2780 in addition to above mentioned cancer cells. The compounds 8–13 also showed 50% inhibition below 10 µg/mL. Furthermore, at concentration of 1 µg/mL, bromophenolics 10, 11 and 12 induced a 77.5, 71.4 and 80.1% inhibition ratio of protein tyrosine kinase (PTK) with the overexpression of c-kit, which was identified in certain malignant diseases and presents a novel approach in cancer therapy [109].
Bis(2,3-dibromo-4,5-dihydroxybenzyl) ether 12 (BDDE), was isolated from the ethanolic extract of red alga, Rhodomela confervoides, alongside with three other bromophenols 15–17. Structurally, all these compounds possess a single phenolic ring. Their cytotoxic activity was investigated against three human cancer cell lines, keratin-forming tumor cell line HeLa (KB), Bel7402 and A549, as well as normal, embryo lung fibroblasts (HELF). Compound 15 exhibited the strongest and most selective anticancer activity with IC50 values of 3.09, 3.18 and 3.54 µg/mL against KB, Bel7402 and A549 cells, respectively. Interestingly, bromophenol 17 induced stronger inhibition of proliferation against normal, HELF cells, while BDDE (12) was selectively active against KB cells only [110]. Liu and colleagues [111] synthesized bromophenol 12 and observed a potent and selective inhibitory activity against several types of cancer cells with determined IC50 values below 40 µg/mL. Compound 12 was the most active against human myelogenous leukemia cells (K562) since it inhibited 50% of proliferation at a concentration of 13.9 µg/mL [112]. The authors also performed a mechanistic study, which revealed that treatment with BDDE 12 modulated the level of pro- and anti-apoptotic proteins Bax and Bcl-2, respectively as well as caspases-3 and -9, indicating the induction of mitochondrial pathway-related apoptosis in K562 cells. The data obtained from the DNA relaxation assay, intercalation assay and molecular docking suggested that BDDE 12 acts as a topoisomerase I inhibitor by binding in the minor groove rather than intercalating into DNA [112]. However, since another study by Liu’s group [113] showed that 12 can intercalate in DNA, further evaluation is needed. The same group highlighted the potential use of compound 12 in cancer therapy, targeting angiogenesis since treatment with human endothelial cells (HUVEC) caused a decrease in the expression of VEGF and its receptor (VEGFR), associated with in vivo effects on zebrafish embryos, more specifically by inhibiting subintestinal vessel formation [114].
Marine algae, L. nana and R. confervoides, are natural sources of bis (2,3-dibromo-4,5-dihydroxy-phenyl)-methane (14) (BDDPM), which was extracted and synthesized by Wu and co-workers [115]. As reported by the authors, compound 14 exhibited a significant anti-proliferative activity against HeLa, RKO, HCT116, Bel7402 and U87 human cancer cell lines with IC50 values: 17.63, 11.37, 10.58, 8.7 and 23.69 µg/mL, respectively. Further studies using hepatoma cells (Bel7402) indicated that BDDPM 14 stimulated cell morphology change, mitochondrial-related apoptosis associated with the cleavages of caspases 3 and 9 and PARP, together with the inhibition of cell migration. The latter was concluded based on reduced wound healing and the inhibition of β1-integrin, known for mediating signal transduction between the cell and ECM. The authors suggested that 14 could be further developed into a novel anti-metastatic agent [115]. Similarly to BDDE 12, BDDPM 14 inhibited angiogenesis in HUVEC cells which resulted from a broad inhibition of several receptor tyrosine kinases and reduced the production of NO which was proven to promote cancer cell invasiveness [116].
Other interesting bromophenol compounds are lanosol (18) and their derivatives, lanosol methyl ether (19), lanosol n-propyl ether (21) and lanosol aldehyde (22), which were isolated from red alga, Polysiphonia lanosa [117]. Lanosol ethyl ether (20) was later found to be an artefact resulting from fractionation over silica gel and using ethyl acetate, as concluded by Shoeib et al. [118]; however, here is presented the anticancer activity of 20 due to its contribution to the understanding of the structure–activity relationship. The authors screened bromophenols 18–22 against colon cancer cell lines, DLD-1 and HCT-116. Compounds 18–21 induced 50% of growth inhibition in DLD-1 cells at a concentration of 18.3, 14.6, 13.5 and 12.4 µM, respectively. On the other side, IC50 values determined after the treatment of HCT-116 cells with 18–21 were 20.4, 14.1, 2.51 and 1.32 µM, respectively. Therefore, it can be concluded that not only the presence of hydroxyl groups and bromine substituents affected the antitumor activity of the above-mentioned compounds, but also the length of the side chain. An increase in chain length led to a higher potency of the compounds, as can be observed for bromophenols 19 to 21. Lanosol aldehyde 22 was the least active against DLD-1 (IC50 = 30.9 µM) and it was not tested against HCT-116 [118]. In 2009, a novel lanosol derivative, lanosol butanone (23), was isolated from another marine red alga, Osmundaria colensoi, and displayed significant anticancer activity against HL-60 cells (IC50 = 8.0 µM) [119].
Ma and colleagues extracted phenylethanol bromophenol (24), structurally similar to lanosol (18), and three unusual derivatives of 24, bearing a sulfate group 25–27 also from the ethanolic extract of R. confervoides [120]. The authors reported moderate inhibitory activity of 24–27 against A549, A2780, BGC-823, Bel7402 and HCT-8 cancer cells, wherein the determined IC50 values were mostly in the range 12-21 µM. Notably, bromophenol sulfate (25) displayed the lowest IC50 value of 9.4 µM against human ovarian cancer [120].
In addition to the red and brown algae, the green species were also used for the isolation of interesting bromophenolic compounds. For example, the tropical green algae, Avrainvillea nigricans, was used by Colon and his team to isolate 3-bromo-4,5-dihydroxybenzyl alcohol (28). At a concentration of 8.9 µg/mL, 28 induced 50% inhibition of proliferation of KB cells, indicating moderate cytostatic activity. Structurally, the compound 28 differs from lanosol (18) in additional bromide substituent in ortho position to the benzylic alcohol [121]. Recently, extraction of another Avrainvillea species, A. amadelpha, resulted in the isolation of avrainvilleal (29) which exhibited moderate activity against HeLa cancer cells (IC50 = 9.64 ± 1.7 µM) [122]. On the other side, Wegener and Miller reported the first total synthesis of avrainvilleol, a similar compound to 29, containing benzyl alcohol group [123].
2.3. Flavonoids
Flavonoids 30–34 (Figure 3) are characterized as a class of polyphenols with polyhydroxylated 2-phenylchromen-4-one backbone [124]. Except the simplest, penimethavone A (30), all the other members of this group 31–34 are monoglycosylated with glucose or galactose. Interestingly, marine-derived flavonoids are known to contain unusual substituents, including the methyl, chlorine, amino and sulfate groups. The latter are considered to be the result of ecological adaptation [125].
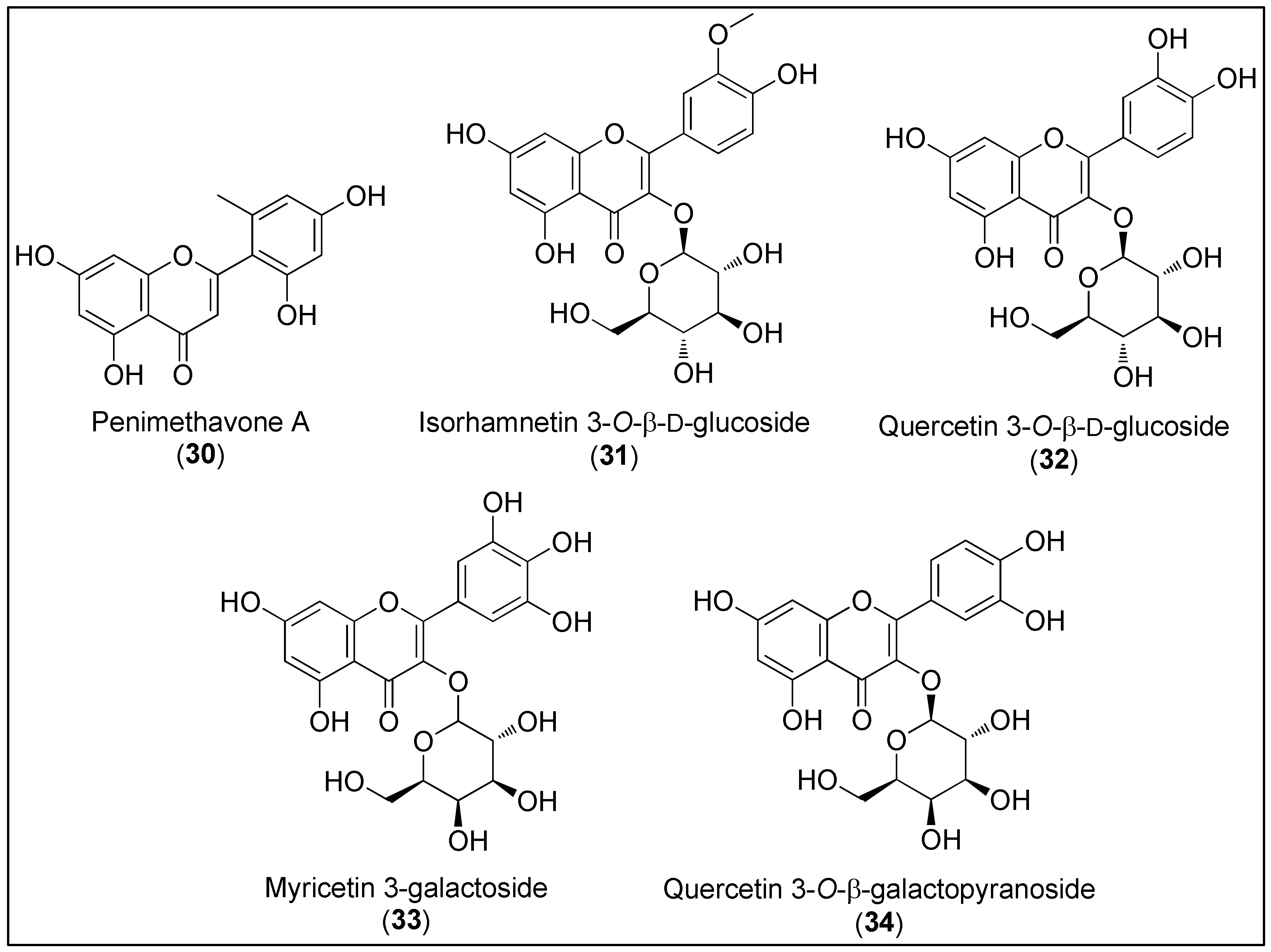
Figure 3. Chemical structures of flavonoids, penimethavone A (30), isolated from fungus Penicillium chrysogenum and flavonoid glycosides 31–34 isolated from the halophytes, Salicornia herbacea and Limonium tetragonum.
Compared to terrestrial flavonoids, marine congeners are noticeably less isolated, characterized and evaluated regarding biological activities [1][125][126]. As stated in an exhaustive review by Martins and co-workers [125], less than 100 flavonoids have been isolated, mostly from seagrasses and halophyte. However, molluscs, bacteria, fungi and corals were also reported as potential natural sources of these secondary metabolites. Since marine flavonoids have been acquainted to exhibit a broad spectrum of biological effects, such as antioxidant, antifouling, antitumor, antimicrobial and antidiabetic, these chemicals present a potential hit or lead compounds in the pharmaceutical industry. Martins et al. also emphasized that only a few dozen of flavonoids have been screened for biological activities [125].
Hou et al. [127] isolated a novel and unusual flavone, penimethavone A (30), from the fungus Penicillium chrysogenum derived from gorgonian coral Carijoa sp. The spectroscopic analysis revealed the presence of a methyl substituent at phenyl ring B, which is quite a rare group at this position among natural compounds. Shao’s group evaluated anticancer activity of 30 against human laryngeal epithelial cancer (Hep-2), A549, HeLa and rhabdomyosarcoma cells (RD). The last two cell lines were the most affected by penimethavone (30) with reported IC50 values of 8.41 and 8.18 µM, respectively [127].
Flavonoid glycosides, isorhamnetin-3-O-β-ᴅ-glucoside (31) and quercetin-3-O-β-ᴅ-glucoside (32), were isolated from a halophyte plant, Salicornia herbacea. Based on the results of Kong and co-workers, the authors suggested that compounds 31 and 32 can be used as chemopreventive agents [128]. The inhibitory activities of 31 and 32 on MMP-2 and MMP-9 in HT1080 cells, associated with an increase in TIMP-1 protein, which is an endogenous inhibitor of the aforementioned MMPs, were observed. A further mechanistic approach revealed the possible connection between the downregulation of MMP genes and suppressed AP-1 promoter activity [128]. Moreover, the cytostatic effect of 32 against HCT116 cells was reported (IC50 = 24.3 μg/mL) [129]. As stated by Mohammed et al., anticancer activity of flavonoid glycosides might be affected by the presence of sugar moiety and the number of hydroxyl groups, both increasing the hydrophilicity of metabolites and, additionally, making an internalization into cells difficult [129].
In 2017, a bioassay-guided fractionation of crude Limonium tetragonum extract resulted in the isolation of two known flavonoid glycosides, myricetin 3-galactoside (33) and quercetin 3-O-β-ᴅ-galactopyranoside (34). Both compounds 33 and 34 exerted inhibitory activities against MMPs in an HT1080 cell line in a selective manner. As observed for flavonoids 31 and 32, the compounds 33 and 34 also suppressed the overexpression of MMP-2 and MMP-9 while simultaneously elevating TIMP-1 and TIMP-2 at the mRNA and protein levels. Compared to 33, the mechanistic studies considering the MAPK signalling pathway revealed that compound 34 significantly reduces the levels of phosphorylated ERK and p-38 [130].
2.4. Coumarins
Coumarins 35–41 present a large and heterogenous class of naturally occurring phenolic compounds which can be found in higher plants, bacteria and fungi, as well as other marine organisms such as molluscs, tunicates or sponges (Figure 4) [131][132]. Structurally, they are characterized by a benzo-α-pyrone skeleton in which the aromatic is connected to a lactone ring [133]. Their role as secondary metabolites is to the protect from the herbivores, microorganisms and infection via antioxidative and enzyme inhibitory activities [131]. Numerous scientific studies have demonstrated miscellaneous pharmacological effects of coumarins, including antioxidative, cardioprotective, anti-inflammatory and anticancer activities [132][134]. The latter arises from their ability to inhibit the protein kinase and telomerase, induce a caspase-mediating apoptosis and exert an anti-angiogenic effect, as discussed by Önder [131]. Due to the simplicity of a benzopyrone skeleton accompanied with the possibility to easily obtain structural diversity by varying the degree of oxygenation, coumarins have attracted much attention from the medicinal chemists in order to develop new structural analogues for therapeutic utilization [135][136].
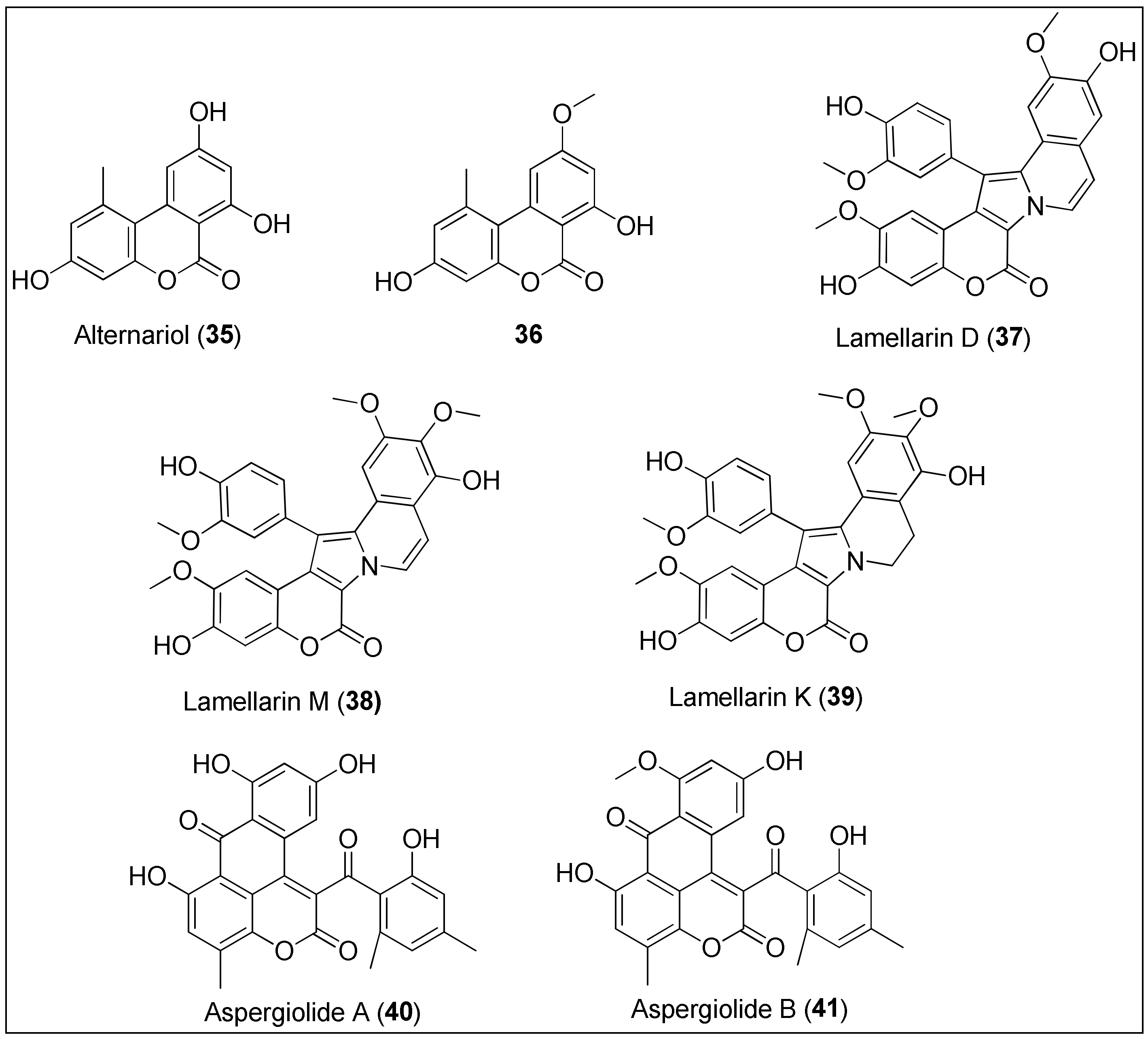
Figure 4. Chemical structures of coumarins: alternariol (35) and its analogue 36 isolated from the mangrove endophytic fungus 2240. Lamellarins D (37), M (38) and K (39) are obtained from the marine molluscs of the genus Lamellaria. Aspergiolides A (40) and B (41) are extracted from the marine fungus, Aspergillus galucus.
Tan et al. [137] isolated alternariol (35) and its derivatives from the mangrove endophytic fungus 2240 and evaluated their antiproliferative potential by means of an MTT assay against two epidermoid carcinoma cell lines, KB and KBv200, of which the latter is a multidrug resistant cell line. Besides alternariol (35), only the alternariol methyl ether (36) exhibited significant activity with IC50 values being 3.17, 3.12 and 4.82, 4.94 μg/mL for KB and KBv200, respectively. Interestingly, the authors observed a positive correlation between a number of hydroxyl groups and a strength of antitumor effect [137]. Both 35 and 36 were also isolated from the fungus, Alternaria alterata, residing in the soft coral, Litophyton arboreum. Alternariol (35) displayed anticancer properties against leukemia cell lines (L1210 and CCRF-CEM), while derivative 36 was active against colon and lung cancer cells (Colon-38 and H-125, respectively). However, both benzocoumarins 35 and 36 were toxic to the normal human cells, the granulocyte–macrophage progenitor (CFU-GM) [138].
Lamellarins are among the most interesting marine coumarins with a wide variety of biological activities. Lamellarins are characterized by a 14-phenyl-6H-[139] benzopyrano[40′,3′:4,5]pyrrolo[2,1-α] isoquinolin-6-one ring system and, because of the central pyrrole ring, can be also considered as alkaloids. Since 1985, more than 50 lamellarins have been discovered from various marine organisms. Lamellarins D (37), M (38) and K (39) are among the most cytotoxic, exhibiting IC50 values often in a nanomolar concentration range against different cancer cell lines [140][141]. For example, lamellarin D (37) was isolated from the marine molluscs of the genus Lamellaria and assayed against 12 human cancer cells, of which LNCaP, DU-145 and K562 were strongly affected, as demonstrated by the 50% growth inhibition concentrations in the 10–20 nM range. Furthermore, murine and human leukemia cells, both sensitive (P-388, CEM) and resistant (P388CPT5, CEM/C2) to camptothecin, respectively, were used for the evaluation of the correlation of topoisomerase I and cytotoxicity. At concentrations of 136 and 1482 nM, 37 inhibited growths of P-388 and P388CPT5 by 50%, respectively, while it was more active against human cell lines with determined IC50 values of 14 and 969 nM against CEM and CEM/C2, respectively. Lamellarin D (37) was found to be a novel topoisomerase I inhibitor due to intercalation into the DNA–topoisomerase I complex, resulting in its stabilization [142]. That was also observed by Ballot and her group [143] who reported another mode of antitumor activity by targeting cancer cell mitochondria [144][145]. As stated by the authors, compound 37 induced apoptosis of the above mentioned cell lines, P388 and topoisomerase I–mutated subclone P388CPT5 by increasing the levels of proapoptotic protein, Bax, and decreasing the expression of antiapoptotic proteins, Bcl-2 and cIAP2, along with caspase-3/-9 activation [143]. The great interest in this compound facilitated total syntheses of 37 and its structural analogues, together with the simultaneous evaluation of their biological activities [146][147]. Furthermore, lamellarins M (38) and K (39), found in the tunicates of the genus Didemnum, remarkably inhibited the growth of several cancer cell lines, more precisely, P388, multidrug-resistant P388 (Schabel), a wild type of chinese hamster ovary cells, CHO (AUXB1), AUXB1 cells resistant to doxorubicin (CCHRC5), A549, HT29, and human melanoma cells (MEL28). The measured 50% inhibitory concentrations were 0.15, 0.17, 0.07, 0.17, 0.06, 0.56, 0.54 and 0.19, 0.017, 0.19, 0.75, 0.18, 0.38, 0.40 μM for 38 and 39, respectively [148]. Recently, some new lamellarins with specific structures and promising anticancer activities were discovered, as discussed by Vazquez-Rodriguez et al. [132].
In 2007, Du and co-workers first reported the isolation, structural characterization and biological evaluation of the anthraquinone derivative, aspergiolide A (40), derived from the marine fungus, Aspergillus galucus. At concentrations of 0.13, 0.28, 7.5 and 35 μM, 40 inhibited the growth of A-549, HL-60, BEL-7402 and P388 cells for 50%, respectively [149]. Considerably more detailed and extensive studies were conducted in 2014, investigating both the anticancer and pharmacokinetic properties of aspergiolide A (40). Compound 40 exhibited significant activity against 11 cancer cells with micromolar IC50 values (2.37–7.07 μM). The Western blot analysis showed that 40 induced caspase-mediated apoptosis of BEL-7402 cancer cells, related to the increase and decrease of Bax and Bcl-2 expressions, respectively. Further, the inhibition of DNA topoisomerase II by 40 was revealed, comparable to the positive control, adriamycin but with less toxic consequences. Finally, compound 40 successfully suppressed the growth of H22 and BEL-7402 cancer xenografts in mice without major effects on body weight [150]. A structural analogue of 40, aspergiolide B (41), was also isolated by the group mentioned above from the same fungus species, A. glaucus. The authors observed that the O-methylation of the hydroxyl group at position C-8 did not affect the anticancer properties of 41 since they reported IC50 values of 0.24 and 0.51 μM against A-549 and HL-60 cancer cell lines, respectively [151]. Thus, the presence of the naphtho[1,2,3-de]chromene-2,7-dione structural feature, characteristic for both aspergiolides A (40) and B (41), might be used for the synthesis of new anticancer derivatives. Moreover, an in silico analysis indicated that 41 is a potential EGFR-TK inhibitor displaying low binding free energy in active site containing MET-766, THR-790 and THR-854 amino acid residues [152].
2.5. Terpenophenolics
The structural complexity of marine secondary metabolites is manifested by the presence of terpenophenolic compounds, in particular, meroditerpenes 42–47 and merosesquiterpenes 48–51 (Figure 5). The first include chromenes, chromanols and plastoquinones which contain a hydroquinone skeleton linked to the side polyprenyl chain. The other hydroquinones will be discussed in the Section 3.6 [153]. The biosynthesis, structural features and biological activities of marine merosesquiterpenes were already extensively reviewed by Le Bideau et al. [154]. Their structures contain a phenolic core derived from the polyketide pathway and a unique, isoprenoid cyclic moiety. Although they can be isolated from marine sponges and gorgonian soft corals [155], brown macroalgae of the genus Stypopodium [156][157][158][159] and red seagrasses of the genus Laurbncia [160][161][162][163] are the main producers of these molecules. Like other marine-derived compounds, terpenophenolics differ in oxygenation and unsaturation levels as well as in the presence of one or more halogen atoms (Figure 5). Many studies have demonstrated their broad-spectrum of biological activities including ichthyotoxicity, insecticidal activity, tyrosine kinase inhibition, antimicrobial activity, microtubule inhibition and the antiproliferative effect against cancer cells [153][154][156][157][159].
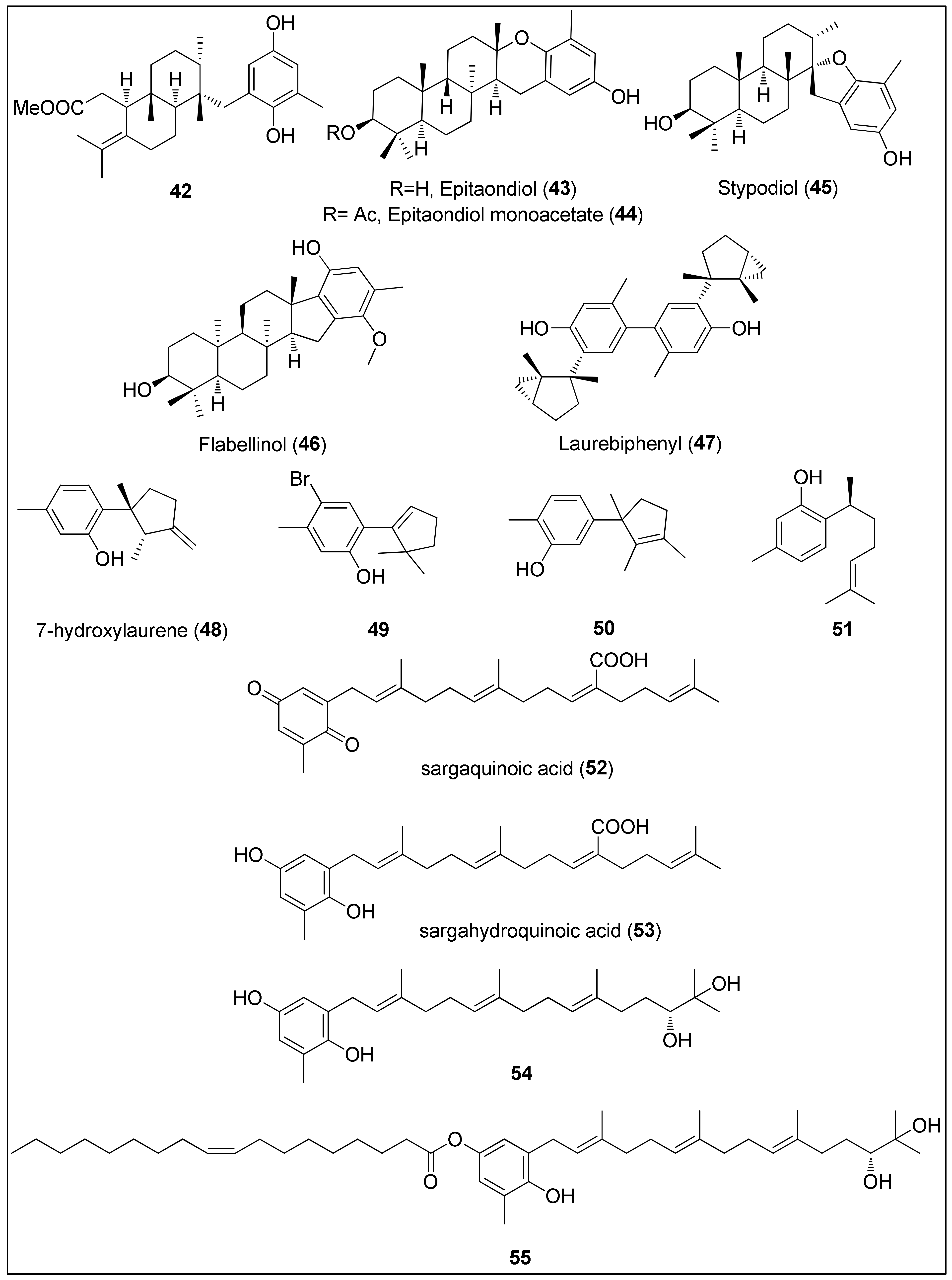
Figure 5. Chemical structures of meroditerpenes 42–47 isolated from brown algae (Stypopodium spp.). Sesquiterpenes 48–50 isolated from red algae (Laurencia spp.), 51 isolated from sponge, Didiscus flavus, and plastoquinones 52–55 isolated from brown algae Sargassum sp.
Dorta and his colleagues used a brown alga, Stypopodium zonale from Macaronesia Archipelago, to extract terpenoids bearing phenolic moiety. Compound 42 displayed potent activity against HT-29, H-116 and A549 cells with IC50 values being 2.5 μg/mL or less [156]. Two other studies, using a different species of brown algae, Stypopodium flabelliforme, resulted in the isolation of the meroditerpenoids 43–46 [157][159]. Their anticancer activities were evaluated against several cancer and non-transformed cell lines by Pereira et al. [157]. The highest activity of almost 100% inhibition of cell proliferation was observed against human neuroblastoma cells (SH-SY5Y) for all compounds. Even though the authors did not provide the exact IC50 values, they can be clearly seen from the reported graphical representations as 6.25–12.5, <12.5 and 12.5–25 μM for epitaondiol (43), epitaondiol monoacetate (44) and stypodiol (45), respectively. Since the only difference between 43 and 44 is the absence and presence of the acetoxy group, it can be assumed that acetylation of 43 decreased biological activities. Interestingly, 43 and 44 were remarkably active against Chinese hamster fibroblasts (V79), unlike stypodiol (45), which showed little effect on normal cells [157]. The neurotoxicity of meroditerpenes from S. flabelliforme was also demonstrated by Sabry and co-workers [159]. They assayed flabellinol (46) against mouse neuroblastoma cell lines (Neuro-2a) which displayed LC50 values in the range from 2 to 11 μM. Moreover, at a concentration of 9 μM, 46 inhibited the growth by 50% of NCI-H460 cancer cells [159].
Shizuri and Yamada were first to isolate and determine the structure of dimeric sesquiterpene, laurebiphenyl (47), from the red algae, Laurencia nidifica, comprising of a unique, cyclolaurane-type of the skeleton [161]. Compound 47 was later extracted from Laurencia tristricha, collected from the Naozhou Island and tested against several cancer cell lines. Sun and co-workers [160] reported significant cytotoxicity of laurebiphenyl (47) against BGC-823, HeLa, A549, HCT-8 and Bel7402 cells with determined IC50 values of 1.22, 1.61, 1.68, 1.77 and 1.91 μg/mL, respectively. Roussis and his team used another Laurencia species, L. microcladia from the North Aegean Sea, to obtain novel cytotoxic sesquiterpenes 48–50, which were published in two independent studies [162][163]. 7-Hydroxylaurene (48) exhibited an antiproliferative effect against human cancer cell lines (MCF-7, PC3, A431, HeLa and K562) as well as CHO, inducing 50% of proliferation inhibition at concentrations of 15.8, 18.1, 23.9, 40.5, 64.2 and 78.2 μM, respectively. As hypothesized by the authors, the presence of an exocyclic-methylene group might be responsible for the observed biological activity [162]. Finally, sesquiterpenes 49 and sterochemically undefined 50 exhibited mild antiproliferative activities against lung cancer cells, NSCLC-N6 (IC50 = 73.4 and 83.7 μM) and A549 (IC50 = 52.4 and 81.0 μM), respectively. The authors assumed that the presence of the hydroxyl group and the unsaturated cyclopentenyl moiety led to higher activity while the bromine did not remarkably affect the overall results [163].
Another bisabolane-type sesquiterpene phenol, (+)-curcuphenol (51), was firstly isolated from the marine sponge, Didiscus flavus, in 1987 by Wright et al. [155]. The compound 51 was also found in another marine sponge, Myrmekioderma styx, as well as a terrestrial plant, Baccharis genistelloides [164]. At a concentration of 7 μg/mL, 51 induced 50% inhibition of P-388 cells, while the minimum inhibitory concentrations against A-549, MDA-MB and HCT-8 were determined as 10, 0.1 and 0.1 μg/mL, respectively [164].
Further, 51 demonstrated moderate antiproliferative activity against four HCT-116 cells (with or without the expression of the p53 and p21 genes). Its activity did not depend on the p53 nor p21 mechanism since IC50 values of 27, 33, 33 and 35 μg/mL were determined for p53+/+, p53−/−, p21+/+ and p21−/−, respectively [165]. Finally, Rodrigo and her colleagues used CaCo-2 colon cancer cells to evaluate the action mechanism of (+)-curcuphenol (51). They showed that this secondary metabolite inhibited the proliferation and DNA synthesis associated with the induction of apoptosis via caspase-3 activation [164].
The organic extract of an Australian marine brown alga, Sargassum fallax, was used to isolate plastoquinones, sargaquinoic acid (52) and sargahydroquinoic acid (53). Both 52 and 53 exhibited prominent antitumor activity with IC50 values of 17 and 14 μM against P388 cells, respectively [166]. A previous study of Hur and colleagues had revealed that sargaquinoic acid (52) induced caspase-mediated apoptosis in a human keratinocyte cell line, HaCaT, while it had no effect on the Bcl-2 and Bax proteins’ expressions [167]. On the other side, plastoquinones 54 and 55 obtained from another Sargassum species, S. micracanthum, displayed significantly higher and comparable antiproliferative activity against the murine colon 26-L5 adenocarcinoma cell line (IC50 = 1.51 and 1.69 μg/mL, respectively) [168].
2.6. Quinones and Hydroquinones
Quinones possess a conjugated cyclic dione function and hydroquinones are their reduced derivatives. They belong to the aromatic organic compounds and can be obtained by the oxidation processes of certain phenolic molecules. They differ in carbon skeletons and might be formed by inter- and intramolecular cyclizations further linked to specific amino acids residues or carbohydrate units. In addition, quinone and hydroquinone moieties can also be present in terpenes and terpenoids, of which some were discussed in the previous section (vide supra) that resulted in a rather challenging classification [169][170]. Chemical structures of quinones and hidroquinones originating from marine species, mainly Streptomyces sp., are presented in Figure 6 and Figure 7.
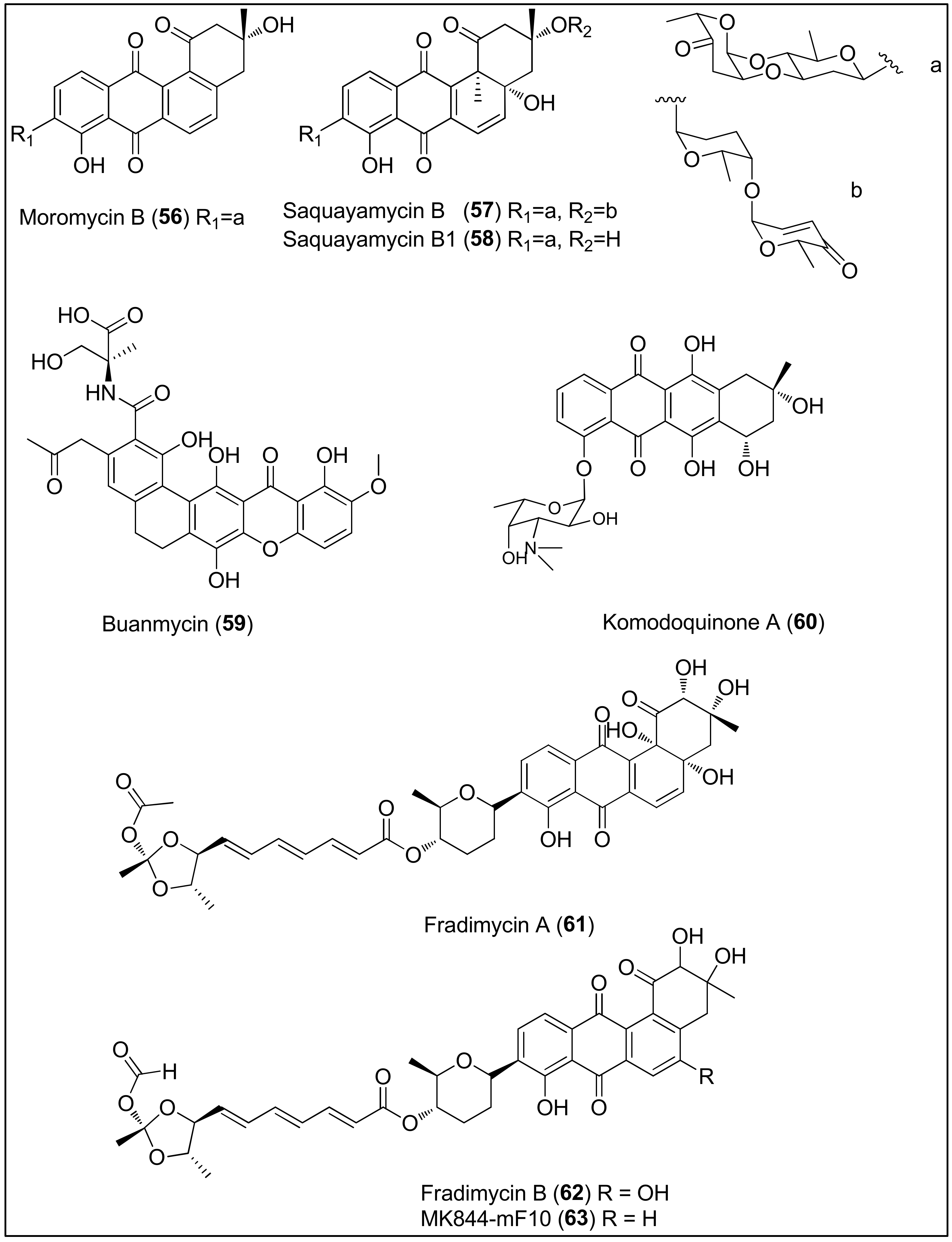
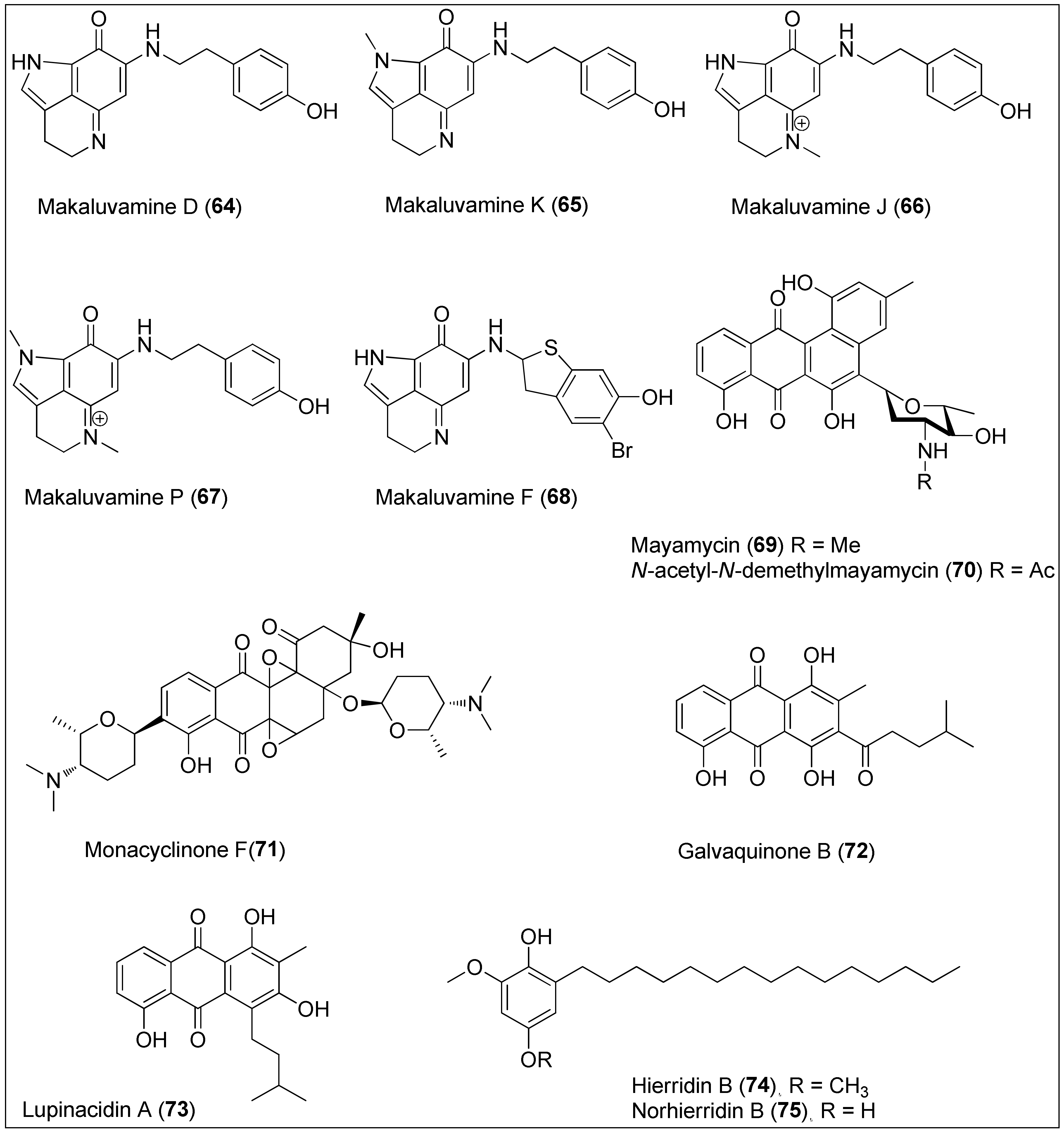
Figure 6. Chemical structures of moromycin B (56), saquayamycins B (57) and B1 (58) isolated from the Streptomyces sp. OC1610.4 strain, buanmycin (59), isolated from the Streptomyces strain, SNR69, komodoquinone A (60), isolated from the Streptomyces sp. KS3 strain and fradimycin A (61), fradimycin B (62) and MK844-mF10 (63), isolated from the Streptomyces fradiae strain PTZ0025.
Figure 7. Makaluvamines D (64), K (65), J (66), P (67) and F (68) isolated from the two sponge genera of Zyzzya and Latrunculi, mayamycin (69) and N-acetyl-N-demethylmayamycin (70) isolated from the Streptomyces sp. strains HB202 and 182SMLY, respectively; monacyclinone F (71) isolated from the Streptomyces sp. strain M7_15, galvaquinone B (72) and lupinacidin A (73) isolated from the Streptomyces spinoverrucosus and hierridin B (74) isolated from the Phormidium ectocarpi and Cyanobium sp. and its structural analogue, norhierrdin B (75).
The broth medium of the marine-derived Streptomyces sp. OC1610.4 strain was used to extract angucycline glycosides, moromycin B (56), saquayamycins B (57) and B1 (58), consisting of tetrangomycin core C- or O-linked to one or two deoxysugar units (Figure 6). Cytotoxicity assays revealed that all three compounds, 56–58, remarkably reduced the proliferation of breast cancer cells (MCF-7, MDA-MB-231 and BT-474) in a sub-micromolar range (0.16–0.67 μM), comparable to the standard control, doxorubicin. Furthermore, transwell and wound-healing assays showed potential antimetastatic properties of 57 since it reduced the invasion and migration of MDA-MB-231 cells at concentrations of 25 and 50 nM [171].
Another marine Streptomyces strain, SNR69, which was later found to be the most similar to Streptomyces cyaneus, was collected from a tidal mud in Buan (Republic of Korea) and identified as a producer of novel, pentacyclic buanmycin (59) that exerted both antibacterial and cytotoxic properties. As reported by the authors, 59 strongly inhibited 50% of the proliferation of A549, HCT116, SNU638, SK-HEP1 and MDA-MB-231 cells at concentrations of 1.7, 0.9, 0.8, 1.9 and 1.2 μM, respectively, similarly to etoposide used as a positive control. Contrary, 59 showed no activity towards the K562 cell line [172].
Itoh and co-workers isolated and determined the absolute stereochemistry of komodoquinone A (60) from the marine Streptomyces sp. KS3 strain. It is an anthracycline containing an amino sugar connected to the ring system and bearing a unique methyl substituent at position 9. As shown, compound 60 can induce morphological changes and lead to neuritogenic activity against the neuroblastoma cell line (Neuro 2A). Interestingly, the cell cycle of Neuro 2A cells was arrested at the G1 phase, contrary to other anthracycline antibiotics which intercalate in DNA, indicating a different mechanism of action of 60. The authors assumed that the carbohydrate moiety might be a prerequisite for the observed biological results since the aglycon part was weakly active [173][174].
In 2012, a study performed by Xin and colleagues using the marine Streptomyces fradiae strain, PTZ0025, led to the isolation of the capoamycin-type antibiotics 61, 62 and 63, characterized by a benz[a]anthraquinone core linked to a deoxysugar and polyenyl side chain [175]. Moreover, another Streptomyces fradiae strain, BDMS1, was also found to produce the abovementioned metabolites [176]. As reported by the authors, fradimycin A (61), fradimycin B (62) and analogue MK844-mF10 (63) exhibited potent inhibitory activity in vitro, against human colon cancer cells (HCT-15 and SW620) and rat glioma cells (C6). Compound 62 was the most active, displaying IC50 values of 0.13, 4.33 and 0.47 μM against HCT-15, SW620 and C6, respectively. For that reason, the mechanism of action of compound 62 was further studied and it was found that 62 induced cell cycle arrest at the G0/G1 phase associated with an increase of apoptotic and necrotic cells [175].
Many studies were performed on interesting pyrroloiminoquinone alkaloids containing a makaluvamine-type scaffold and phenolic substituent, which were isolated from the two sponge genera of Zyzzya and Latrunculia collected in the Pacific–Oceania region. In addition, a significant cytotoxicity of makaluvamines has been reported through interaction with topoisomerase II resulting in DNA cleavage [177][178]. Among them, makaluvamines D (64), K (65), J (66) and P (67) were found to be the most active against the PANC-1 cell line, exhibiting IC50 values of 0.29, 0.56, 0.054 and 0.3 μM, respectively. Structure–activity relationship studies revealed three main structural properties responsible for potent anticancer activity: a conjugation system in the main makaluvamine core, the presence of a cationic tetrahydropyridinium moiety, and a tyramineyl substituent. Furthermore, since makaluvamine J (66) also had an IC50 value in a nanomolar range (120 nM) against the ovarian cancer cell line OVCAR-5, the authors decided to continue with preclinical studies by using 66 [179]. Despite the existence of a planar structure and a positive charge that contributes to the high affinity for DNA, already in 2005, Dijoux and her colleagues showed that intercalation into DNA is not the only mode of its action [180]. On the other side, structurally different and stereochemically undefined makaluvamine F (68) also displayed strong inhibitory activity in a sub-micromolar range against HCT-116 with a determined IC50 value of 0.17 μM [181]. Further, in 2016, Goey et al. found that this natural product reduced the activity of HIF-1α and its downstream target, VEGF indicating the possible role of makaluvamines in hypoxia conditions [182]. Synthetic approaches in the preparation of some makaluvamines, as well as their structural analogues, were also reported [183][184][185].
The culture broth of the Streptomyces sp. strain HB202, derived from the marine sponge, Halichondria panicea, was used to isolate and characterize mayamycin (69), a novel benz[a]anthracene derivative. More precisely, 69 is known for its unique C-bounded angolosamine unit, with a dimethylamino group at the C-5 of the skeleton. Mayamycin (69) exhibited potent activity with determined IC50 values of 0.2, 0.3, 0.2, 0.16, 0.29, 0.13, 0.15 and 0.33 μM against eight cancer cell lines, HepG2, HT-29, GXF251L, LXF529L, MAXF401NL, MEXF462NL, PAXF1657L and RXF486L, respectively. As published by the authors, 69 was also cytotoxic toward a mouse fibroblast cell line (NIH-3T3) [186]. An analogue of 69, N-acetyl-N-demethylmayamycin (70), was obtained in 2016 from another marine Streptomyces strain, 182SMLY, by Liang et al. [187]. The authors reported that treatment with 70 resulted in 50% of proliferation inhibition at the concentrations of 0.7, 1.4, 3.9 and 0.5 μM, against U251, U87-MG, SHG-44 and C6 glioma cell lines, respectively, as determined by sulforhodamine B assay. Furthermore, it was shown that 70 can induce apoptosis in U251 cells [187]. Synthetic routes toward mayamycin have been discussed and published emphasizing intramolecular aldol condensation and Hauser annulation as key steps [188][189].
In 2015, six new angucyclinone derivatives were extracted from the sponge-derived Streptomyces sp. Strain, M7_15, of which monacyclinone F (71) showed the highest activity against rhabdomycosarcoma cancer cells (SJCRH30) displaying an EC50 value of 0.73 μM. The authors concluded that the structural characteristic arising from the presence of two epoxide rings, aminodeoxysugar and ketone moiety could be of great importance for biological activity [190].
Anthraquinones with alkyl substituents, galvaquinone B (72) and lupinacidin A (73), were isolated from the marine-derived Streptomyces spinoverrucosus by Hu and colleagues [191]. They screened both metabolites for cytotoxic activity against Calu-3 and H2887 cancer cell lines and reported IC50 values of 5.0 and 12.2 μM for 72 and 8.8 and 3.1 μM for 73, respectively. Recently, Sottorff and his team isolated both 72 and 73 from the sea anemone (Gyractis sesere) from Easter Island, but further confirmed the Actinobacteria of the genus Verrucosispora as the exact producer of those compounds [192].
In 1998, Papendorf and his colleagues used the marine cyanobacterium Phormidium ectocarpi to isolate hierrdin B (74), a methylated hydrouinone with a long aliphatic chain which showed antiplasmodial activity [193]. However, its cytotoxic potential was examined against a panel of human cancer cell lines only in 2013. Leão et al. purified 74 from the marine picocyanobacterium Cyanobium sp. LEGE 06113, and determined its selective, but weak, activity towards HT-29 cells with an IC50 value of 100.2 µM [194]. The same group used the aforementioned cells to evaluate the mechanism underlying the biological activity of hierrdin B (74). The authors pointed out that 74 targets mitochondrial activity by increasing the mRNA expression of VDAC1, a key protein involved in mitochondria-mediated apoptosis. That observation was accompanied with the inhibition of cell cycle progression induced by 74 [195]. In the meantime, an analogue of the natural product 74, norhierridin B (75), was synthesized with improved inhibitory activity against several cancer cell lines by activating the p53 pathway. The IC50 values of 0.61, 0.77, 0.68, 2.0, 0.61 and 3.2 µM against MDA-MB-231, SKBR3, MDA-MB-468, A375, Huh-7 and HCT116 were measured, respectively. Therefore, the authors suggested that the presence of two hydroxy groups in the quinone skeleton is of great importance for the improvement of anticancer activity [196].
2.7. Miscellaneous Compounds
The ethyl-acetate extract of the sponge-derived fungus of the genus Didymellaceae was used to isolate several phenol derivatives including diorcinol L (76) (Figure 8). It exhibited potent antiproliferative activity against Huh-7, DU145, HeLa and HL60 cancer cell lines with determined IC50 values of 5.7, 9.1, 7.1 and 9.6 µM, respectively [197]. Interestingly, strong activity against cancer cells including DU145 and HeLa of 76, isolated from the endophytic algae-derived fungus, Aspergillus tennesseensis, was not observed by Zhang et al. However, the authors reported that the dihydrobenzofuran derivative 77 displayed activity towards THP-1 cells since it inhibited the 50% growth of the cells at the concentration of 7.0 µg/mL [198]. On the other side, the benzophenone derivative, sulochrin (78), a compound somewhat similar to diorcinol L (76), was very recently isolated from the Red Sea-derived fungus, Aspergillus falconensis [199]. At a concentration of 5.1 µM, 78 inhibited 50% growth of a mouse lymphoma cell line (L5178Y), while MDA-MB-231 cell migration was inhibited at 70 µM. The authors also performed docking studies which revealed inhibition activity of 78 against the CDK-2, TOP-2 and MMP-13 proteins.
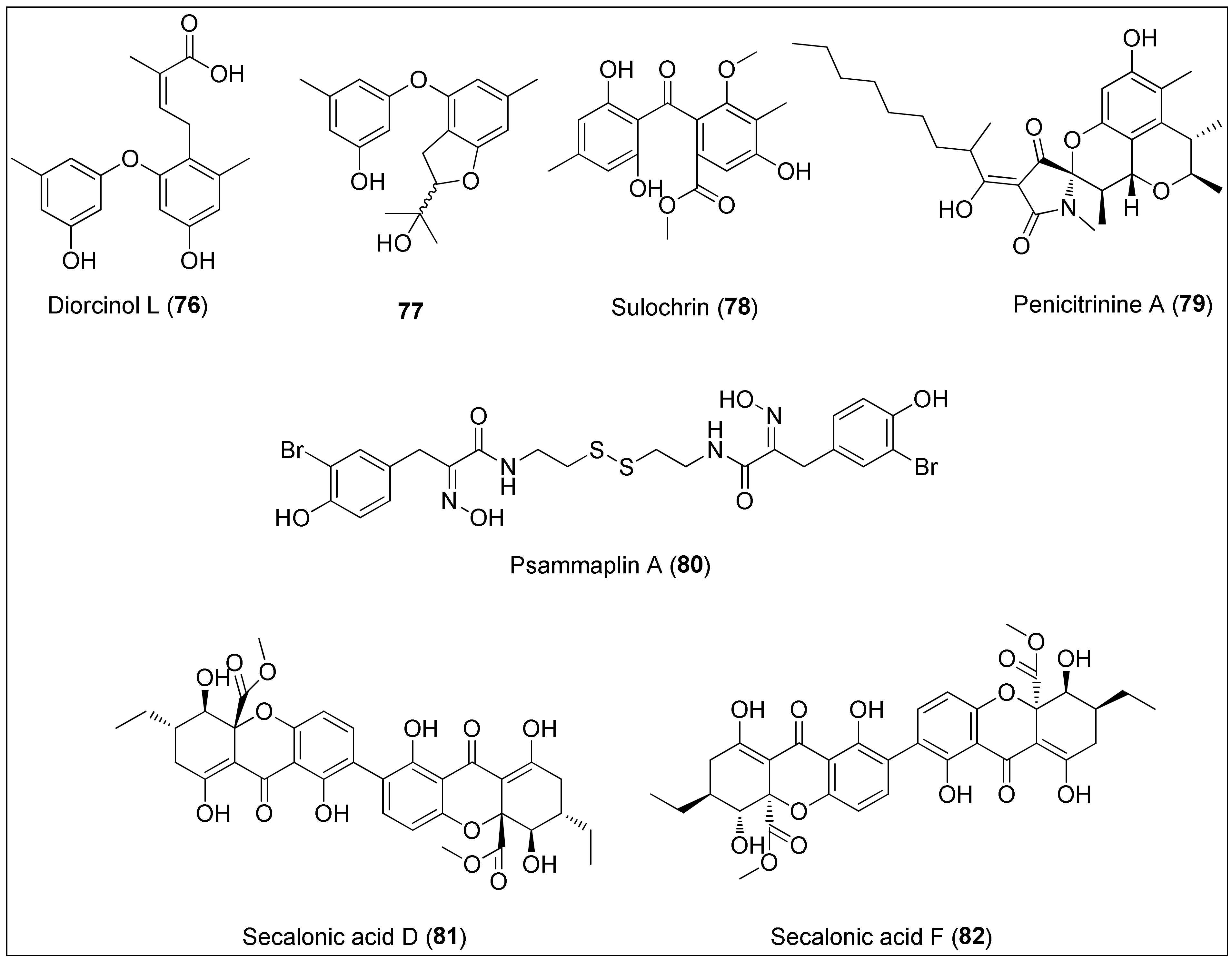
Figure 8. Diorcinol L (76) isolated from the fungus of the genus Didymellaceae and its derivative 77, sulochrin (78) obtained from the fungus Aspergillus falconensis, penicitrinine A (79) isolated from the Penicillium citrinum, psammaplin A (80) extracted from the marine sponge of the genus Pseudoceratina, and secalonic acids D (81) and F (82) obtained from Penicillium sp. and Aspergillus aculeatus.
Penicitrinine A (79) is a phenolic derivative with a unique spiro skeleton that was isolated from the marine fungus Penicillium citrinum. It showed a promising and moderate activity against various solid tumor types in vitro, with A-375 cells being the most sensitive. At the concentrations of 30.88, 12.78 and 7.06 µM, a treatment with 79 for 24 h, 48 h and 72 h, respectively, resulted in 50% growth inhibition of the abovementioned cell line. Furthermore, Liu and co-workers investigated the mechanism of action in detail regarding apoptotic and metastatic activity. The authors revealed that penicitrinine A (79) induced apoptosis of A-375 cells by decreasing and increasing the expression of the Bcl-2 and Bax proteins, respectively. Additionally, 79 induced cell migration suppression by downregulating MMP-9 and upregulating TIMP-1 levels [200].
An unusual bromotyrosine metabolite containing oxime and disulfide moieties, psammaplin A (80), was isolated from the marine sponge of the genus Pseudoceratina [201]. Several studies have demonstrated anticancer effects of this disulfide dimer, exerted both in vitro and in vivo, as recently extensively reviewed by Jing and co-workers [201]. In brief, the cytotoxicity of 80 is manifested in the regulation of the expression of proteins involved in angiogenesis, DNA replication, apoptosis, proliferation and invasion. Therefore, it has been revealed that psammaplin A (80) inhibits aminopeptidase N (APN), mycothiol-S-conjugate amidase (MCA), topoisomerase II, farnesyl protein transferase, histone deacetylases (HDACs) and leucine aminopeptidase [201]. More importantly, synthetic approaches were developed to prepare both 80 and its analogues, which were found as more potent inhibitors of HDACs [202] and DOT1L (disruptor of telomeric silencing-1 like) than the natural product 80 [203].
Tang et al. used the culture extract of the marine-derived Penicillium oxalicum to extract secalonic acid D (81). The authors demonstrated selective cytotoxic effects of 81 towards PANC-1 cells (IC50 value of 0.6 µM) which were adapted to certain nutrient conditions associated with the assumption of the mechanism of action via the inhibition of the Akt signalling pathway [204]. Furthermore, 81 displayed potent cytotoxicity against both sensitive and multidrug resistant cells with determined IC50 values being: 6.8, 6.4, 5.3, 4.9, 5.1 and 4.9 µM against S1, S1-MI-80, H460, H460/MX20, MCF-7 and MCF-7/ADR, respectively. In addition, 81 was found to induce cell death through c-Jun/Src/STAT3 signalling by inhibiting the proteasome-dependent degradation of c-Jun [205]. Another research, performed by Guru and colleagues, used secalonic acid D (81), however, isolated from the terrestrial source. It was revealed that 81 exhibited antitumor activity in both normal (HUVEC) and MCF-7 cancer cells through the Akt/mTOR/p70S6K pathway resulting in the inhibition of eNOS and ERK phosphorylation together with MMP degradation as key pro-angiogenesis factors [206].
On the other hand, an isomer of 81, secalonic acid F (82), has been isolated from the marine-derived fungal strains, Penicillium sp. F11 and Aspergillus aculeatus [207][208]. A study by Li and colleagues [208] revealed 50% of growth inhibition of HL-60 cells at the concentration of 4.1 µg/mL, further associated with induction of apoptosis via caspase-3 activation and the modulation of the RhoGDI2 protein. The latter is connected to the invasion and metastasis, thus being recognized as a novel therapeutic approach in cancer treatment [209]. Further, a phenolic derivative 82 was more potent towards HepG2 cells than the positive control (5-FU), exhibiting IC50 values of: 45.5, 8.7 and 7.7 µM after 24, 48 and 72 h treatment, respectively. Those observations were, in relation to mitochondrial-mediated apoptosis, more precisely the activation of caspases-3 and -9. The authors also performed in vivo studies that demonstrated lower tumor weights after treatment with 82 [207]. A new biological evaluation of secalonic acid F (82) resulted in the identification of a potential target, MARCH1, for the treatment of hepatocellular carcinoma, in which the downregulation suppressed the migration and invasion of HepG2 and Hep3B cancer cells [210]. Finally, the most recent study obtained by Özenver and co-workers demonstrated the potency and selective toxicity of 82 against leukemia and multiple myeloma cells with regard to normal cells mediated through apoptosis and necrosis, as well as tubulin disassembly [211].
References
- Cotas, J.; Leandro, A.; Monteiro, P.; Pacheco, D.; Figueirinha, A.; Goncąlves, A.M.M.; Da Silva, G.J.; Pereira, L. Seaweed phenolics: From extraction to applications. Mar. Drugs 2020, 18, 384.
- Mekinić, I.G.; Skroza, D.; Šimat, V.; Hamed, I.; Čagalj, M.; Perković, Z.P. Phenolic content of brown algae (Pheophyceae) species: Extraction, identification, and quantification. Biomolecules 2019, 9, 244.
- Utkina, N.K.; Makarchenko, A.E.; Shchelokova, O.V.; Virovaya, M.V. Antioxidant activity of phenolic metabolites from marine sponges. Chem. Nat. Compd. 2004, 40, 373–377.
- Cichewicz, R.H.; Clifford, L.J.; Lassen, P.R.; Cao, X.; Freedman, T.B.; Nafie, L.A.; Deschamps, J.D.; Kenyon, V.A.; Flanary, J.R.; Holman, T.R.; et al. Stereochemical determination and bioactivity assessment of (S)-(+)-curcuphenol dimers isolated from the marine sponge Didiscus aceratus and synthesized through laccase biocatalysis. Bioorganic Med. Chem. 2005, 13, 5600–5612.
- Putra, M.Y.; Murniasih, T.; Swasono, R.T.; Wibowo, J.T.; Saputri, A.N.C.; Widhiana, M.R.; Arlyza, I.S. Secondary metabolites and their biological activities in Indonesian soft coral of the genus Lobophytum. Asian Pac. J. Trop. Biomed. 2016, 6, 909–913.
- Deghrigue, M.; Dellai, A.; Akremi, N.; Le Morvan, V.; Robert, J.; Bouraoui, A. Evaluation of antiproliferative and antioxidant activities of the organic extract and its polar fractions from the Mediterranean gorgonian Eunicella singularis. Environ. Toxicol. Pharmacol. 2013, 36, 339–346.
- Deghrigue, M.; Dellai, A.; Bouraoui, A.; Akremi, N.; Le Morvan, V.; Robert, J.; Bouraoui, A. In Vitro Antiproliferative and Antioxidant Activities of the Organic Extract and Its Semi-Purified Fractions from the Mediterranean Gorgonian Eunicella singularis. Int. J. Pharm. Pharm. Sci. 2013, 5, 432–439.
- Matulja, D.; Grbčić, P.; Bojanić, K.; Topić-Popović, N.; Čož-Rakovac, R.; Laclef, S.; Šmuc, T.; Jović, O.; Marković, D.; Pavelić, S.K. Chemical evaluation, antioxidant, antiproliferative, anti-inflammatory and antibacterial activities of organic extract and semi-purified fractions of the Adriatic sea fan, Eunicella cavolini. Molecules 2021, 26, 5751.
- Hussein, H.A.; Abdullah, M.A. Anticancer Compounds Derived from Marine Diatoms. Mar. Drugs 2020, 18, 356.
- Zheng, Y.; Chen, X.; Chen, L.; Shen, L.; Fu, X.; Chen, Q.; Chen, M.; Wang, C. Isolation and Neuroprotective Activity of Phenolic Derivatives from the Marine-Derived Fungus Penicillium janthinellum. J. Ocean Univ. China 2020, 19, 700–706.
- Kodzius, R.; Gojobori, T. Marine metagenomics as a source for bioprospecting. Mar. Genomics 2015, 24, 21–30.
- Metsämuuronen, S.; Sirén, H. Bioactive phenolic compounds, metabolism and properties: A review on valuable chemical compounds in Scots pine and Norway spruce. Phytochem. Rev. 2019, 18, 623–664.
- Bertelli, A.; Biagi, M.; Corsini, M.; Baini, G.; Cappellucci, G.; Miraldi, E. Polyphenols: From Theory to Practice. Foods 2021, 10, 2595.
- Mageroy, M.H.; Jancsik, S.; Saint Yuen, M.M.; Fischer, M.; Withers, S.G.; Paetz, C.; Schneider, B.; Mackay, J.; Bohlmann, J. A conifer UDP-sugar dependent glycosyltransferase contributes to acetophenone metabolism and defense against insects. Plant Physiol. 2017, 175, 641–651.
- Mateos, R.; Pérez-Correa, J.R.; Domínguez, H. Bioactive properties of marine phenolics. Mar. Drugs 2020, 18, 501.
- Vacca, R.A.; Valenti, D.; Caccamese, S.; Daglia, M.; Braidy, N.; Nabavi, S.M. Plant polyphenols as natural drugs for the management of Down syndrome and related disorders. Neurosci. Biobehav. Rev. 2016, 71, 865–877.
- Taskin, D.; Ozdemir, M.; Yalcin, B. LC-ESI-tandem MS and in silico ADMET analysis of polyphenols from Rhus coriaria L. and Micromeria fruticosa (L.) Druce ssp. brachycalyx P. H. Davis. Futur. J. Pharm. Sci. 2021, 7, 168.
- Getachew, A.T.; Jacobsen, C.; Holdt, S.L. Emerging technologies for the extraction of marine phenolics: Opportunities and challenges. Mar. Drugs 2020, 18, 389.
- Bhatia, P.; Chugh, A. Role of marine bioprospecting contracts in developing access and benefit sharing mechanism for marine traditional knowledge holders in the pharmaceutical industry. Glob. Ecol. Conserv. 2015, 3, 176–187.
- Berni, R.; Cai, G.; Hausman, J.F.; Guerriero, G. Plant fibers and phenolics: A review on their synthesis, analysis and combined use for biomaterials with new properties. Fibers 2019, 7, 80.
- Stewart, A.J.; Stewart, R.F. Phenols. In Encyclopedia of Ecology; Jørgensen, S.E., Fath, B.D., Eds.; Academic Press: Oxford, UK, 2008; pp. 2682–2689. ISBN 9780080914565.
- Anantharaju, P.G.; Gowda, P.C.; Vimalambike, M.G.; Madhunapantula, S.V. An overview on the role of dietary phenolics for the treatment of cancers. Nutr. J. 2016, 15, 99.
- Saltveit, M.E. Synthesis and metabolism of phenolic compounds. In Fruit and Vegetable Phytochemicals: Chemistry and Human Health; Yahia, E.M., Ed.; John Wiley & Sons Ltd.: Hoboken, NJ, USA, 2017; pp. 115–124. ISBN 9781119158042.
- Francenia Santos-Sánchez, N.; Salas-Coronado, R.; Hernández-Carlos, B.; Villanueva-Cañongo, C. Shikimic Acid Pathway in Biosynthesis of Phenolic Compounds. In Plant Physiological Aspects of Phenolic Compounds; Soto-Hernández, S., García-Mateos, R., PalmaTenango, M., Eds.; IntechOpen: London, UK, 2019; Chapter 3; pp. 35–50.
- Tohge, T.; De Souza, L.P.; Fernie, A.R. Current understanding of the pathways of flavonoid biosynthesis in model and crop plants. J. Exp. Bot. 2017, 68, 4013–4028.
- Babenko, L.M.; Smirnov, O.E.; Romanenko, K.O.; Trunova, O.K.; Kosakivska, I.V. Phenolic compounds in plants: Biogenesis and functions. Ukr. Biochem. J. 2019, 91, 5–18.
- Kumari, S.; Priya, P.; Misra, G.; Yadav, G. Structural and biochemical perspectives in plant isoprenoid biosynthesis. Phytochem. Rev. 2013, 12, 255–291.
- Mandrekar, V.K.; Gawas, U.B.; Majik, M.S. Brominated Molecules From Marine Algae and Their Pharmacological Importance. In Studies in Natural Products Chemistry; Atta-ur-Rahman, Ed.; Elsevier B.V.: Amsterdam, The Netherlands, 2018; Volume 61, pp. 461–490. ISBN 978-0-444-64183-0.
- Bertoni, G. A key step in phlorotannin biosynthesis revealed. Plant Cell 2013, 25, 2770.
- Meslet-Cladière, L.; Delage, L.; Leroux, C.J.J.; Goulitquer, S.; Leblanc, C.; Creis, E.; Gall, E.A.; Stiger-Pouvreau, V.; Czjzek, M.; Potin, P. Structure/function analysis of a type III polyketide synthase in the brown alga Ectocarpus siliculosus reveals a biochemical pathway in phlorotannin monomer biosynthesis. Plant Cell 2013, 25, 3089–3103.
- Jimenez-Lopez, C.; Pereira, A.G.; Lourenço-Lopes, C.; Garcia-Oliveira, P.; Cassani, L.; Fraga-Corral, M.; Prieto, M.A.; Simal-Gandara, J. Main bioactive phenolic compounds in marine algae and their mechanisms of action supporting potential health benefits. Food Chem. 2021, 341, 128262.
- Di Meo, F.; Lemaur, V.; Cornil, J.; Lazzaroni, R.; Duroux, J.L.; Olivier, Y.; Trouillas, P. Free radical scavenging by natural polyphenols: Atom versus electron transfer. J. Phys. Chem. A 2013, 117, 2082–2092.
- Aggarwal, V.; Tuli, H.S.; Varol, A.; Thakral, F.; Yerer, M.B.; Sak, K.; Varol, M.; Jain, A.; Khan, M.A.; Sethi, G. Role of reactive oxygen species in cancer progression: Molecular mechanisms and recent advancements. Biomolecules 2019, 9, 735.
- Guedes, É.A.C.; Da Silva, T.G.; Aguiar, J.S.; De Barros, L.D.; Pinotti, L.M.; Sant’Ana, A.E.G. Cytotoxic activity of marine algae against cancerous cells. Brazilian J. Pharmacogn. 2013, 23, 668–673.
- Abu-Khudir, R.; Ismail, G.A.; Diab, T. Antimicrobial, Antioxidant, and Anti-Tumor Activities of Sargassum linearifolium and Cystoseira crinita from Egyptian Mediterranean Coast. Nutr. Cancer 2021, 73, 829–844.
- Namvar, F.; Baharara, J.; Mahdi, A.A. Antioxidant and anticancer activities of selected Persian gulf algae. Indian J. Clin. Biochem. 2014, 29, 13–20.
- Mhadhebi, L.; Mhadhebi, A.; Robert, J.; Bouraoui, A. Antioxidant, anti-inflammatory and antiproliferative effects of aqueous extracts of three Mediterranean brown seaweeds of the Genus Cystoseira. Iran. J. Pharm. Res. 2014, 13, 207–220.
- Premarathna, A.D.; Ranahewa, T.H.; Wijesekera, S.K.; Harishchandra, D.L.; Karunathilake, K.J.K.; Waduge, R.N.; Wijesundara, R.R.M.K.K.; Jayasooriya, A.P.; Wijewardana, V.; Rajapakse, R.P.V.J. Preliminary screening of the aqueous extracts of twenty-three different seaweed species in Sri Lanka with in-vitro and in-vivo assays. Heliyon 2020, 6, e03918.
- Abdelhamid, A.; Lajili, S.; Elkaibi, M.A.; Ben Salem, Y.; Abdelhamid, A.; Muller, C.D.; Majdoub, H.; Kraiem, J.; Bouraoui, A. Optimized Extraction, Preliminary Characterization and Evaluation of the in Vitro Anticancer Activity of Phlorotannin-Rich Fraction from the Brown Seaweed, Cystoseira sedoides. J. Aquat. Food Prod. Technol. 2019, 28, 892–909.
- Zenthoefer, M.; Geisen, U.; Hofmann-Peiker, K.; Fuhrmann, M.; Kerber, J.; Kirchhöfer, R.; Hennig, S.; Peipp, M.; Geyer, R.; Piker, L.; et al. Isolation of polyphenols with anticancer activity from the Baltic Sea brown seaweed Fucus vesiculosus using bioassay-guided fractionation. J. Appl. Phycol. 2017, 29, 2021–2037.
- Park, C.; Lee, H.; Hwangbo, H.; Ji, S.Y.; Kim, M.Y.; Kim, S.Y.; Hong, S.H.; Kim, G.Y.; Choi, Y.H. Ethanol extract of Hizikia fusiforme induces apoptosis in B16F10 mouse melanoma cells through ROS-dependent inhibition of the PI3K/Akt signaling pathway. Asian Pacific J. Cancer Prev. 2020, 21, 1275–1282.
- Sevimli-Gur, C.; Yesil-Celiktas, O. Cytotoxicity screening of supercritical fluid extracted seaweeds and phenylpropanoids. Mol. Biol. Rep. 2019, 46, 3691–3699.
- Kosanić, M.; Ranković, B.; Stanojković, T. Brown macroalgae from the Adriatic Sea as a promising source of bioactive nutrients. J. Food Meas. Charact. 2019, 13, 330–338.
- Rosa, G.P.; Tavares, W.R.; Sousa, P.M.C.; Pagès, A.K.; Seca, A.M.L.; Pinto, D.C.G.A. Seaweed secondary metabolites with beneficial health effects: An overview of successes in in vivo studies and clinical trials. Mar. Drugs 2020, 18, 8.
- Imbs, T.I.; Zvyagintseva, T.N. Phlorotannins are Polyphenolic Metabolites of Brown Algae. Russ. J. Mar. Biol. 2018, 44, 263–273.
- Erpel, F.; Mateos, R.; Pérez-Jiménez, J.; Pérez-Correa, J.R. Phlorotannins: From isolation and structural characterization, to the evaluation of their antidiabetic and anticancer potential. Food Res. Int. 2020, 137, 109589.
- Singh, I.P.; Sidana, J. Phlorotannins. In Functional Ingredients from Algae for Foods and Nutraceuticals; Domínguez, H., Ed.; Woodhead Publishing: Cambridge, UK, 2013; pp. 181–204. ISBN 9780857095121.
- Nahvi, I.; Belkahla, S.; Asiri, S.M.; Rehman, S. Overview and Prospectus of Algal Biogenesis of Nanoparticles. In Microbial Nanotechnology: Green Synthesis and Applications; Ansari, M.A., Rehman, S., Eds.; Springer: Singapore, 2021; pp. 121–134. ISBN 978-981-16-1923-6.
- Bai, Y.; Sun, Y.; Gu, Y.; Zheng, J.; Yu, C.; Qi, H. Preparation, Characterization and Antioxidant Activities of Kelp Phlorotannin Nanoparticles. Molecules 2020, 25, 4550.
- Kaushalya, K.G.D.; Gunathilake, K.D.P.P. Encapsulation of phlorotannins from edible brown seaweed in chitosan: Effect of fortification on bioactivity and stability in functional foods. Food Chem. 2022, 377, 132012.
- Singh, I.P.; Sidana, J.; Bharate, S.B.; Foley, W.J. Phloroglucinol compounds of natural origin: Synthetic aspects. Nat. Prod. Rep. 2010, 27, 393–416.
- Pal Singh, I.; Bharate, S.B. Phloroglucinol compounds of natural origin. Nat. Prod. Rep. 2006, 23, 558–591.
- Kang, K.A.; Zhang, R.; Chae, S.; Lee, S.J.; Kim, J.; Kim, J.; Jeong, J.; Lee, J.; Shin, T.; Lee, N.H.; et al. Phloroglucinol (1,3,5-trihydroxybenzene) protects against ionizing radiation-induced cell damage through inhibition of oxidative stress in vitro and in vivo. Chem. Biol. Interact. 2010, 185, 215–226.
- Kwon, Y.H.; Jung, S.Y.; Kim, J.W.; Lee, S.H.; Lee, J.H.; Lee, B.Y.; Kwon, S.M. Phloroglucinol inhibits the bioactivities of endothelial progenitor cells and suppresses tumor angiogenesis in LLC-tumor-bearing mice. PLoS ONE 2012, 7, e33618.
- Kim, R.K.; Suh, Y.; Yoo, K.C.; Cui, Y.H.; Hwang, E.; Kim, H.J.; Kang, J.S.; Kim, M.J.; Lee, Y.Y.; Lee, S.J. Phloroglucinol suppresses metastatic ability of breast cancer cells by inhibition of epithelial-mesenchymal cell transition. Cancer Sci. 2015, 106, 94–101.
- Kang, M.H.; Kim, I.H.; Nam, T.J. Phloroglucinol induces apoptosis via apoptotic signaling pathways in HT-29 colon cancer cells. Oncol. Rep. 2014, 32, 1341–1346.
- Kang, M.H.; Kim, I.H.; Nam, T.J. Phloroglucinol induces apoptosis through the regulation of insulin-like growth factor 1 receptor signaling pathways in human colon cancer HT-29 cells. Int. J. Oncol. 2014, 45, 1036–1042.
- Kim, R.K.; Uddin, N.; Hyun, J.W.; Kim, C.; Suh, Y.; Lee, S.J. Novel anticancer activity of phloroglucinol against breast cancer stem-like cells. Toxicol. Appl. Pharmacol. 2015, 286, 143–150.
- Lopes-Costa, E.; Abreu, M.; Gargiulo, D.; Rocha, E.; Ramos, A.A. Anticancer effects of seaweed compounds fucoxanthin and phloroglucinol, alone and in combination with 5-fluorouracil in colon cells. J. Toxicol. Environ. Health—Part A Curr. Issues 2017, 80, 1–12.
- Kang, K.A.; Lee, K.H.; Chae, S.; Zhang, R.; Jung, M.S.; Lee, Y.; Kim, S.Y.; Kim, H.S.; Joo, H.G.; Park, J.W.; et al. Eckol isolated from Ecklonia cava attenuates oxidative stress induced cell damage in lung fibroblast cells. FEBS Lett. 2005, 579, 6295–6304.
- Park, E.; Ahn, G.N.; Lee, N.H.; Kim, J.M.; Yun, J.S.; Hyun, J.W.; Jeon, Y.J.; Wie, M.B.; Lee, Y.J.; Park, J.W.; et al. Radioprotective properties of eckol against ionizing radiation in mice. FEBS Lett. 2008, 582, 925–930.
- Hyun, K.H.; Yoon, C.H.; Kim, R.K.; Lim, E.J.; An, S.; Park, M.J.; Hyun, J.W.; Suh, Y.; Kim, M.J.; Lee, S.J. Eckol suppresses maintenance of stemness and malignancies in glioma stem-like cells. Toxicol. Appl. Pharmacol. 2011, 254, 32–40.
- Zhang, M.; Zhou, W.; Zhao, S.; Li, S.; Yan, D.; Wang, J. Eckol inhibits Reg3A-induced proliferation of human SW1990 pancreatic cancer cells. Exp. Ther. Med. 2019, 18, 2825–2832.
- Zhu, Y.; Guo, J.; Hu, X.; Liu, J.; Li, S.; Wang, J. Eckol protects against acute experimental colitis in mice: Possible involvement of Reg3g. J. Funct. Foods 2020, 73, 104088.
- Zhang, M.Y.; Guo, J.; Hu, X.M.; Zhao, S.Q.; Li, S.L.; Wang, J. An in vivo anti-tumor effect of eckol from marine brown algae by improving the immune response. Food Funct. 2019, 10, 4361–4371.
- Cho, S.H.; Kim, H.S.; Lee, W.W.; Han, E.J.; Kim, S.Y.; Fernando, I.P.S.; Ahn, G.; Kim, K.N. Eckol from Ecklonia cava ameliorates TNF-α/IFN-γ-induced inflammatory responses via regulating MAPKs and NF-κB signaling pathway in HaCaT cells. Int. Immunopharmacol. 2020, 82, 106146.
- Eo, H.J.; Kwon, T.H.; Park, G.H.; Song, H.M.; Lee, S.J.; Park, N.H.; Jeong, J.B. In vitro anticancer activity of phlorofucofuroeckol a via upregulation of activating transcription factor 3 against human colorectal cancer cells. Mar. Drugs 2016, 14, 69.
- Lee, Y.J.; Park, J.H.; Park, S.A.; Joo, N.R.; Lee, B.H.; Lee, K.B.; Oh, S.M. Dieckol or phlorofucofuroeckol extracted from Ecklonia cava suppresses lipopolysaccharide-mediated human breast cancer cell migration and invasion. J. Appl. Phycol. 2020, 32, 631–640.
- Manandhar, B.; Wagle, A.; Seong, S.H.; Paudel, P.; Kim, H.R.; Jung, H.A.; Choi, J.S. Phlorotannins with potential anti-tyrosinase and antioxidant activity isolated from the marine seaweed Ecklonia stolonifera. Antioxidants 2019, 8, 240.
- Zhang, C.; Li, Y.; Qian, Z.J.; Lee, S.H.; Li, Y.X.; Kim, S. Dieckol from Ecklonia cava regulates invasion of human fibrosarcoma cells and modulates mmp-2 and mmp-9 expression via NF-b pathway. Evid.-Based Complement. Altern. Med. 2011, 2011, 140462.
- Oh, S.M.; Park, C.G.; Kang, J.H.; Kim, E.J.; Chee, H.Y.; Lee, B.H.; Lee, K.B. Dieckol inhibits 12-O-tetradecanoylphorbol-13-acetate-induced SK-Hep1 human hepatoma cell motility through suppression of matrix metalloproteinase-9 activity. J. Appl. Biol. Chem. 2011, 54, 376–381.
- Wang, L.; Lee, W.; Jayawardena, T.U.; Cha, S.-H.; Jeon, Y.-J. Dieckol, an algae-derived phenolic compound, suppresses airborne particulate matter-induced skin aging by inhibiting the expressions of pro-inflammatory cytokines and matrix metalloproteinases through regulating NF-κB, AP-1, and MAPKs signaling pathways. Food Chem. Toxicol. 2020, 146, 111823.
- Park, S.J.; Kim, Y.T.; Jeon, Y.J. Antioxidant dieckol downregulates the Rac1/ROS signaling pathway and inhibits Wiskott-Aldrich syndrome protein (WASP)-family verprolin-homologous protein 2 (WAVE2)-mediated invasive migration of B16 mouse melanoma cells. Mol. Cells 2012, 33, 363–369.
- Park, S.J.; Jeon, Y.J. Dieckol from Ecklonia cava suppresses the migration and invasion of HT1080 cells by inhibiting the focal adhesion kinase pathway downstream of Rac1-ROS signaling. Mol. Cells 2012, 33, 141–149.
- Takenawa, T.; Miki, H. WASP and WAVE family proteins: Key molecules for rapid rearrangement of cortical actin filaments and cell movement. J. Cell Sci. 2001, 114, 1801–1809.
- Yoon, J.S.; Kasin Yadunandam, A.; Kim, S.J.; Woo, H.C.; Kim, H.R.; Kim, G. Do Dieckol, isolated from Ecklonia stolonifera, induces apoptosis in human hepatocellular carcinoma Hep3B cells. J. Nat. Med. 2013, 67, 519–527.
- Li, Y.X.; Li, Y.; Je, J.Y.; Kim, S.K. Dieckol as a novel anti-proliferative and anti-angiogenic agent and computational anti-angiogenic activity evaluation. Environ. Toxicol. Pharmacol. 2015, 39, 259–270.
- Ahn, J.H.; Yang, Y.I.; Lee, K.T.; Choi, J.H. Dieckol, isolated from the edible brown algae Ecklonia cava, induces apoptosis of ovarian cancer cells and inhibits tumor xenograft growth. J. Cancer Res. Clin. Oncol. 2014, 141, 255–268.
- Kim, E.K.; Tang, Y.; Kim, Y.S.; Hwang, J.W.; Choi, E.J.; Lee, J.H.; Lee, S.H.; Jeon, Y.U.J.; Park, P.J. First evidence that Ecklonia cava-derived dieckol attenuates MCF-7 human breast carcinoma cell migration. Mar. Drugs 2015, 13, 1785–1797.
- You, S.H.; Kim, J.-S.; Kim, Y.-S. Apoptosis and Cell Cycle Arrest in Two Human Breast Cancer Cell Lines by Dieckol Isolated from Ecklonia cava. J. Breast Dis. 2018, 6, 39–45.
- Sadeeshkumar, V.; Duraikannu, A.; Ravichandran, S.; Kodisundaram, P.; Fredrick, W.S.; Gobalakrishnan, R. Modulatory efficacy of dieckol on xenobiotic-metabolizing enzymes, cell proliferation, apoptosis, invasion and angiogenesis during NDEA-induced rat hepatocarcinogenesis. Mol. Cell. Biochem. 2017, 433, 195–204.
- Sadeeshkumar, V.; Duraikannu, A.; Ravichandran, S.; Fredrick, W.S.; Sivaperumal, R.; Kodisundaram, P. Protective effects of dieckol on N-nitrosodiethylamine induced hepatocarcinogenesis in rats. Biomed. Pharmacother. 2016, 84, 1810–1819.
- Wang, C.H.; Li, X.F.; Jin, L.F.; Zhao, Y.; Zhu, G.J.; Shen, W.Z. Dieckol inhibits non-small–cell lung cancer cell proliferation and migration by regulating the PI3K/AKT signaling pathway. J. Biochem. Mol. Toxicol. 2019, 33, e22346.
- Xu, J.W.; Yan, Y.; Wang, L.; Wu, D.; Ye, N.K.; Chen, S.H.; Li, F. Marine bioactive compound dieckol induces apoptosis and inhibits the growth of human pancreatic cancer cells PANC-1. J. Biochem. Mol. Toxicol. 2021, 35, e22648.
- Park, E.; Ahn, G.; Yun, J.S.; Kim, M.J.; Bing, S.J.; Kim, D.S.; Lee, J.; Lee, N.H.; Park, J.W.; Jee, Y. Dieckol rescues mice from lethal irradiation by accelerating hemopoiesis and curtailing immunosuppression. Int. J. Radiat. Biol. 2010, 86, 848–859.
- Sadeeshkumar, V.; Duraikannu, A.; Aishwarya, T.; Jayaram, P.; Ravichandran, S.; Ganeshamurthy, R. Radioprotective efficacy of dieckol against gamma radiation-induced cellular damage in hepatocyte cells. Naunyn. Schmiedebergs. Arch. Pharmacol. 2019, 392, 1031–1041.
- Toume, K.; Miyata, M.; Egawa, K.; Nose, K.; Hayashi, M.; Komiyama, K.; Ishibashi, M. Isolation of diphlorethohydroxycarmalol from a brown alga Ishige okamurae. Nat. Med. 2004, 58, 79–80.
- Kang, S.M.; Kim, A.D.; Heo, S.J.; Kim, K.N.; Lee, S.H.; Ko, S.C.; Jeon, Y.J. Induction of apoptosis by diphlorethohydroxycarmalol isolated from brown alga, Ishige okamurae. J. Funct. Foods 2012, 4, 433–439.
- Park, M.H.; Jeon, Y.J.; Kim, H.J.; Han, J.S. Effect of diphlorethohydroxycarmalol isolated from Ishige okamurae on apoptosis in 3 t3-L1 preadipocytes. Phyther. Res. 2013, 27, 931–936.
- Piao, M.J.; Kumara, M.H.S.R.; Kim, K.C.; Kang, K.A.; Kang, H.K.; Lee, N.H.; Hyun, J.W. Diphlorethohydroxycarmalol suppresses ultraviolet B-induced matrix metalloproteinases via inhibition of JNK and ERK signaling in human keratinocytes. Biomol. Ther. 2015, 23, 557–563.
- Wang, L.; Kim, H.S.; Oh, J.Y.; Je, J.G.; Jeon, Y.J.; Ryu, B.M. Protective effect of diphlorethohydroxycarmalol isolated from Ishige okamurae against UVB-induced damage in vitro in human dermal fibroblasts and in vivo in zebrafish. Food Chem. Toxicol. 2020, 136, 110963.
- Piao, M.J.; Hewage, S.R.K.M.; Han, X.; Kang, K.A.; Kang, H.K.; Lee, N.H.; Hyun, J.W. Protective effect of diphlorethohydroxycarmalol against ultraviolet B radiation-induced DNA damage by inducing the nucleotide excision repair system in HaCaT human keratinocytes. Mar. Drugs 2015, 13, 5629–5641.
- Park, C.; Lee, H.; Hong, S.H.; Kim, J.H.; Park, S.K.; Jeong, J.W.; Kim, G.Y.; Hyun, J.W.; Yun, S.J.; Kim, B.W.; et al. Protective effect of diphlorethohydroxycarmalol against oxidative stress-induced DNA damage and apoptosis in retinal pigment epithelial cells. Cutan. Ocul. Toxicol. 2019, 38, 298–308.
- Kong, C.S.; Kim, J.A.; Yoon, N.Y.; Kim, S.K. Induction of apoptosis by phloroglucinol derivative from Ecklonia cava in MCF-7 human breast cancer cells. Food Chem. Toxicol. 2009, 47, 1653–1658.
- Yoon, N.Y.; Eom, T.K.; Kim, M.M.; Kim, S.K. Inhibitory effect of phlorotannins isolated from Ecklonia cava on mushroom tyrosinase activity and melanin formation in mouse B16F10 melanoma cells. J. Agric. Food Chem. 2009, 57, 4124–4129.
- Lee, M.S.; Yoon, H.D.; Kim, J.I.; Choi, J.S.; Byun, D.S.; Kim, H.R. Dioxinodehydroeckol inhibits melanin synthesis through PI3K/Akt signalling pathway in α-melanocyte-stimulating hormone-treated B16F10 cells. Exp. Dermatol. 2012, 21, 417–473.
- Ryu, B.M.; Ahn, B.N.; Kang, K.H.; Kim, Y.S.; Li, Y.X.; Kong, C.S.; Kim, S.K.; Kim, D.G. Dioxinodehydroeckol protects human keratinocyte cells from UVB-induced apoptosis modulated by related genes Bax/Bcl-2 and caspase pathway. J. Photochem. Photobiol. B Biol. 2015, 153, 352–357.
- Ham, Y.-M.; Baik, J.-S.; Hyun, J.-W.; Lee, N.-H. Isolation of a new phlorotannin, fucodiphlorethol G, from a brown alga Ecklonia cava. Bull. Korean Chem. Soc. 2007, 28, 1595–1597.
- Li, Y.; Qian, Z.J.; Kim, M.M.; Kim, S.K. Cytotoxic activities of phlorethol and fucophlorethol derivatives isolated from Laminariaceae Ecklonia cava. J. Food Biochem. 2011, 35, 357–369.
- Li, Y.; Qian, Z.J.; Ryu, B.M.; Lee, S.H.; Kim, M.M.; Kim, S.K. Chemical components and its antioxidant properties in vitro: An edible marine brown alga, Ecklonia cava. Bioorganic Med. Chem. 2009, 17, 1963–1973.
- Li, Y.X.; Li, Y.; Qian, Z.J.; Ryu, B.; Kim, S.K. Suppression of vascular endothelial growth factor (VEGF) induced angiogenic responses by fucodiphloroethol G. Process Biochem. 2011, 46, 1095–1103.
- Gribble, G.W. Biological activity of recently discovered halogenated marine natural products. Mar. Drugs 2015, 13, 4044–4136.
- Cavallo, G.; Metrangolo, P.; Milani, R.; Pilati, T.; Priimagi, A.; Resnati, G.; Terraneo, G. The halogen bond. Chem. Rev. 2016, 116, 2478–2601.
- Mantri, R.V.; Sanghvi, R.; Zhu, H.J. Solubility of pharmaceutical solids. In Developing Solid Oral Dosage Forms: Pharmaceutical Theory and Practice, 2nd ed.; Qiu, Y., Chen, Y., Zhang, G.G.Z., Yu, L., Mantri, R.V., Eds.; Elsevier Inc.: Amsterdam, The Netherlands, 2017; pp. 3–22. ISBN 9780128024478.
- Shibata, T.; Miyasaki, T.; Miyake, H.; Tanaka, R.; Kawaguchi, S. The Influence of Phlorotannins and Bromophenols on the Feeding Behavior of Marine Herbivorous Gastropod Turbo cornutus. Am. J. Plant Sci. 2014, 5, 387–392.
- Dong, H.; Dong, S.; Hansen, P.E.; Stagos, D.; Lin, X.; Liu, M. Progress of bromophenols in marine algae from 2011 to 2020: Structure, bioactivities, and applications. Mar. Drugs 2020, 18, 411.
- Shinada, N.K.; De Brevern, A.G.; Schmidtke, P. Halogens in Protein-Ligand Binding Mechanism: A Structural Perspective. J. Med. Chem. 2019, 62, 9341–9356.
- Xu, X.; Song, F.; Wang, S.; Li, S.; Xiao, F.; Zhao, J.; Yang, Y.; Shang, S.; Yang, L.; Shi, J. Dibenzyl bromophenols with diverse dimerization patterns from the brown alga Leathesia nana. J. Nat. Prod. 2004, 67, 1661–1666.
- Shi, D.; Li, J.; Guo, S.; Su, H.; Fan, X. The antitumor effect of bromophenol derivatives in vitro and Leathesia nana extract in vivo. Chinese J. Oceanol. Limnol. 2009, 27, 277–282.
- Lijun, H.; Nianjun, X.; Jiangong, S.; Xiaojun, Y.; Chengkui, Z. Isolation and pharmacological activities of bromophenols from Rhodomela confervoides. Chinese J. Oceanol. Limnol. 2005, 23, 226–229.
- Liu, M.; Zhang, W.; Wei, J.; Lin, X. Synthesis and α-glucosidase inhibitory mechanisms of bis(2,3-dibromo-4,5-dihydroxybenzyl) ether, a potential marine bromophenol α-glucosidase inhibitor. Mar. Drugs 2011, 9, 1554–1565.
- Liu, M.; Zhang, W.; Wei, J.; Qiu, L.; Lin, X. Marine bromophenol bis(2,3-dibromo-4,5-dihydroxybenzyl) ether, induces mitochondrial apoptosis in K562 cells and inhibits topoisomerase I in vitro. Toxicol. Lett. 2012, 211, 126–134.
- Liu, M.; Wang, G.; Xiao, L.; Xu, A.; Liu, X.; Xu, P.; Lin, X. Bis(2,3-dibromo-4,5-dihydroxybenzyl) ether, a marine algae derived bromophenol, inhibits the growth of Botrytis cinerea and interacts with DNA molecules. Mar. Drugs 2014, 12, 3838–3851.
- Qi, X.; Liu, G.; Qiu, L.; Lin, X.; Liu, M. Marine bromophenol bis(2,3-dibromo-4,5-dihydroxybenzyl) ether, represses angiogenesis in HUVEC cells and in zebrafish embryos via inhibiting the VEGF signal systems. Biomed. Pharmacother. 2015, 75, 58–66.
- Wu, N.; Luo, J.; Jiang, B.; Wang, L.; Wang, S.; Wang, C.; Fu, C.; Li, J.; Shi, D. Marine bromophenol bis (2,3-dibromo-4,5-dihydroxy-phenyl)-methane inhibits the proliferation, migration, and invasion of hepatocellular carcinoma cells via modulating β1-integrin/FAK signaling. Mar. Drugs 2015, 13, 1010–1025.
- Wang, S.; Wang, L.J.; Jiang, B.; Wu, N.; Li, X.; Liu, S.; Luo, J.; Shi, D. Anti-angiogenic properties of BDDPM, a bromophenol from marine red alga Rhodomela confervoides, with multi receptor tyrosine kinase inhibition effects. Int. J. Mol. Sci. 2015, 16, 13548–13560.
- Hodgkin, J.H.; Craigie, J.S.; McInnes, A.G. The occurrence of 2,3-dibromobenzyl alcohol 4,5-disulfate, dipotassium salt, in Polysiphonia lanosa. Can. J. Chem. 1966, 44, 74–78.
- Shoeib, N.A.; Bibby, M.C.; Blunden, G.; Linley, P.A.; Swaine, D.J.; Wheelhouse, R.T.; Wright, C.W. In-vitro cytotoxic activities of the major bromophenols of the red alga Polysiphonia lanosa and some novel synthetic isomers. J. Nat. Prod. 2004, 67, 1445–1449.
- Popplewell, W.L.; Northcote, P.T. Colensolide A: A new nitrogenous bromophenol from the New Zealand marine red alga Osmundaria colensoi. Tetrahedron Lett. 2009, 50, 6814–6817.
- Ma, M.; Zhao, J.; Wang, S.; Li, S.; Yang, Y.; Shi, J.; Fan, X.; He, L. Bromophenols coupled with methyl γ-ureidobutyrate and bromophenol sulfates from the red alga Rhodomela confervoides. J. Nat. Prod. 2006, 69, 206–210.
- Colon, M.; Guevara, P.; Gerwick, W.H.; Ballantine, D. 5’-hydroxyisoavrainvilleol, a new Diphenylmethane Derivative from the Tropical Green Alga Avrainvillea nigricans. J. Nat. Prod. 1987, 50, 368–374.
- Hawas, U.W.; Abou El-Kassem, L.T.; Al-Farawati, R.; Shaher, F.M. Halo-phenolic metabolites and their in vitro antioxidant and cytotoxic activities from the Red Sea alga Avrainvillea amadelpha. Zeitschrift fur Naturforsch. 2021, 76, 213–218.
- Wegener, A.; Miller, K.A. Total Synthesis of Avrainvilleol. J. Org. Chem. 2017, 82, 11655–11658.
- Tauchen, J.; Huml, L.; Rimpelova, S.; Jurášek, M. Flavonoids and related members of the aromatic polyketide group in human health and disease: Do they really work? Molecules 2020, 25, 3846.
- Martins, B.T.; Correia da Silva, M.; Pinto, M.; Cidade, H.; Kijjoa, A. Marine natural flavonoids: Chemistry and biological activities. Nat. Prod. Res. 2019, 33, 3260–3272.
- Goiris, K.; Muylaert, K.; Voorspoels, S.; Noten, B.; De Paepe, D.; E Baart, G.J.; De Cooman, L. Detection of flavonoids in microalgae from different evolutionary lineages. J. Phycol. 2014, 50, 483–492.
- Hou, X.M.; Wang, C.Y.; Gu, Y.C.; Shao, C.L. Penimethavone A, a flavone from a gorgonian-derived fungus Penicillium chrysogenum. Nat. Prod. Res. 2016, 30, 2274–2277.
- Kong, C.S.; Kim, Y.A.; Kim, M.M.; Park, J.S.; Kim, J.A.; Kim, S.K.; Lee, B.J.; Nam, T.J.; Seo, Y. Flavonoid glycosides isolated from Salicornia herbacea inhibit matrix metalloproteinase in HT1080 cells. Toxicol. Vitr. 2008, 22, 1742–1748.
- Mohammed, M.M.D.; El-Sharkawy, E.R.; Matloub, A.A. Cytotoxic flavonoids from Diplotaxis harra (Forssk.) Boiss. growing in Sinai. J. Med. Plant Res. 2011, 5, 5099–5103.
- Bae, M.J.; Karadeniz, F.; Oh, J.H.; Yu, G.H.; Jang, M.S.; Nam, K.H.; Seo, Y.; Kong, C.S. MMP-Inhibitory Effects of Flavonoid Glycosides from Edible Medicinal Halophyte Limonium tetragonum. Evid.-Based Complement. Altern. Med. 2017, 2017, 6750274.
- Önder, A. Anticancer activity of natural coumarins for biological targets. In Studies in Natural Products Chemistry; Rahman, A., Ed.; Elsevier: Amsterdam, The Netherlands, 2020; Volume 64, pp. 85–109. ISBN 978-0-12-817903-1.
- Vazquez-Rodriguez, S.; Matos, M.; Borges, F.; Uriarte, E.; Santana, L. Bioactive Coumarins from Marine Sources: Origin, Structural Features and Pharmacological Properties. Curr. Top. Med. Chem. 2015, 15, 1755–1766.
- Matos, M.J.; Santana, L.; Uriarte, E.; Abreu, O.A.; Molina, E.; Yordi, E.G. Coumarins—An Important Class of Phytochemicals. In Phytochemicals—Isolation, Characterisation and Role in Human Health; Rao, A.V., Rao, L.G., Eds.; IntechOpen: London, UK, 2015; pp. 113–140. ISBN 978-953-51-2170-1.
- Shejwalkar, P. Applications of Coumarins as Cardiovascular and Anti-Cancer Agents: A Short Review. J. Cardiol. Cardiovasc. Ther. 2017, 8, 1–6.
- Katsori, A.M.; Hadjipavlou-Litina, D. Coumarin derivatives: An updated patent review (2012–2014). Expert Opin. Ther. Pat. 2014, 24, 1323–1347.
- Detsi, A.; Kontogiorgis, C.; Hadjipavlou-Litina, D. Coumarin derivatives: An updated patent review (2015–2016). Expert Opin. Ther. Pat. 2017, 27, 1201–1226.
- Tan, N.; Tao, Y.; Pan, J.; Wang, S.; Xu, F.; She, Z.; Lin, Y.; Gareth Jones, E.B. Isolation, structure elucidation, and mutagenicity of four alternariol derivatives produced by the mangrove endophytic fungus No. 2240. Chem. Nat. Compd. 2008, 44, 296–300.
- Hawas, U.W.; El-Desouky, S.; Abou El-Kassem, L.; Elkhateeb, W. Alternariol derivatives from Alternaria alternata, an endophytic fungus residing in Red sea soft coral, inhibit HCV NS3/4A protease. Appl. Biochem. Microbiol. 2015, 51, 579–584.
- Torre, L.A.; Bray, F.; Siegel, R.L.; Ferlay, J.; Lortet-Tieulent, J.; Jemal, A. Global cancer statistics, 2012. CA Cancer J. Clin. 2015, 65, 87–108.
- Fukuda, T.; Ishibashi, F.; Iwao, M. Lamellarin alkaloids: Isolation, synthesis, and biological activity. In Alkaloids: Chemistry and Biology; Knölker, H.-J., Ed.; Elsevier Inc.: Amsterdam, The Netherlands, 2020; Volume 83, pp. 1–112. ISBN 9780128209813.
- Bailly, C. Anticancer properties of lamellarins. Mar. Drugs 2015, 13, 1105–1123.
- Facompré, M.; Tardy, C.; Bal-Mahieu, C.; Colson, P.; Perez, C.; Manzanares, I.; Cuevas, C.; Bailly, C. Lamellarin D: A Novel Potent Inhibitor of Topoisomerase I. Cancer Res. 2003, 63, 7392–7399.
- Ballot, C.; Kluza, J.; Martoriati, A.; Nyman, U.; Formstecher, P.; Joseph, B.; Bailly, C.; Marchetti, P. Essential role of mitochondria in apoptosis of cancer cells induced by the marine alkaloid Lamellarin D. Mol. Cancer Ther. 2009, 8, 3307–3317.
- Ballot, C.; Martoriati, A.; Jendoubi, M.; Buche, S.; Formstecher, P.; Mortier, L.; Kluza, J.; Marchetti, P. Another facet to the anticancer response to lamellarin D: Induction of cellular senescence through inhibition of topoisomerase i and intracellular ROS production. Mar. Drugs 2014, 12, 779–798.
- Ballot, C.; Kluza, J.; Lancel, S.; Martoriati, A.; Hassoun, S.M.; Mortier, L.; Vienne, J.C.; Briand, G.; Formstecher, P.; Bailly, C.; et al. Inhibition of mitochondrial respiration mediates apoptosis induced by the anti-tumoral alkaloid lamellarin D. Apoptosis 2010, 15, 769–781.
- Colligs, V.; Hansen, S.P.; Imbri, D.; Seo, E.J.; Kadioglu, O.; Efferth, T.; Opatz, T. Synthesis and biological evaluation of a D-ring-contracted analogue of lamellarin D. Bioorganic Med. Chem. 2017, 25, 6137–6148.
- Lade, D.M.; Pawar, A.B.; Mainkar, P.S.; Chandrasekhar, S. Total Synthesis of Lamellarin D Trimethyl Ether, Lamellarin D, and Lamellarin H. J. Org. Chem. 2017, 82, 4998–5004.
- Quesada, A.R.; García Grávalos, M.D.; Fernández Puentes, J.L. Polyaromatic alkaloids from marine invertebrates as cytotoxic compounds and inhibitors of multidrug resistance caused by P-glycoprotein. Br. J. Cancer 1996, 74, 677–682.
- Du, L.; Zhu, T.; Fang, Y.; Liu, H.; Gu, Q.; Zhu, W. Aspergiolide A, a novel anthraquinone derivative with naphthochromene-2,7-dione skeleton isolated from a marine-derived fungus Aspergillus glaucus. Tetrahedron 2007, 63, 1085–1088.
- Wang, Y.; Qi, X.; Li, D.; Zhu, T.; Mo, X.; Li, J. Anticancer efficacy and absorption, distribution, metabolism, and toxicity studies of aspergiolide A in early drug development. Drug Des. Devel. Ther. 2014, 8, 1965–1977.
- Du, L.; Zhu, T.; Liu, H.; Fang, Y.; Zhu, W.; Gu, Q. Cytotoxic polyketides from a marine-derived fungus Aspergillus glaucus. J. Nat. Prod. 2008, 71, 1837–1842.
- Watty, M.; Syahdi, R.R.; Yanuar, A. Database compilation and virtual screening of secondary metabolites derived from marine fungi as epidermal growth factor receptor tyrosine kinase inhibitors. Asian J. Pharm. Clin. Res. 2017, 10, 142–147.
- De La Mare, J.A.; Lawson, J.C.; Chiwakata, M.T.; Beukes, D.R.; Edkins, A.L.; Blatch, G.L. Quinones and halogenated monoterpenes of algal origin show anti-proliferative effects against breast cancer cells in vitro. Investig. New Drugs 2012, 30, 2187–2200.
- Le Bideau, F.; Kousara, M.; Chen, L.; Wei, L.; Dumas, F. Tricyclic sesquiterpenes from marine origin. Chem. Rev. 2017, 117, 6110–6159.
- Wright, A.E.; Pomponi, S.A.; McConnell, O.J.; Kohmoto, S.; McCarthy, P.J. (+)-Curcuphenol and (+)-curcudiol, sesquiterpene phenols from shallow and deep water collections of the marine sponge Didiscus flavus. J. Nat. Prod. 1987, 50, 976–978.
- Dorta, E.; Cueto, M.; Brito, I.; Darias, J. New terpenoids from the brown alga Stypopodium zonale. J. Nat. Prod. 2002, 65, 1727–1730.
- Pereira, D.M.; Cheel, J.; Areche, C.; San-Martin, A.; Rovirosa, J.; Silva, L.R.; Valentao, P.; Andrade, P.B. Anti-proliferative activity of meroditerpenoids isolated from the brown alga Stypopodium flabelliforme against several cancer cell lines. Mar. Drugs 2011, 9, 852–862.
- Depix, M.S.; Martínez, J.; Santibañez, F.; Rovirosa, J.; Martín, A.S.; Maccioni, R.B. The compound 14-keto stypodiol diacetate from the algae Stypopodium flabelliforme inhibits microtubules and cell proliferation in DU-145 human prostatic cells. Mol. Cell. Biochem. 1998, 187, 191–199.
- Sabry, O.M.M.; Andrews, S.; McPhail, K.L.; Goeger, D.E.; Yokochi, A.; LePage, K.T.; Murray, T.F.; Gerwick, W.H. Neurotoxic meroditerpenoids from the tropical marine brown alga Stypopodium flabelliforme. J. Nat. Prod. 2005, 68, 1022–1030.
- Sun, J.; Shi, D.; Ma, M.; Li, S.; Wang, S.; Han, L.; Yang, Y.; Fan, X.; Shi, J.; He, L. Sesquiterpenes from the red alga Laurencia tristicha. J. Nat. Prod. 2005, 68, 915–919.
- Shizuri, Y.; Yamada, K. Laurebiphenyl, a dimeric sesquiterpene of the cyclolaurane-type from the red alga Laurencia nidifica. Phytochemistry 1985, 24, 1385–1386.
- Kladi, M.; Xenaki, H.; Vagias, C.; Papazafiri, P.; Roussis, V. New cytotoxic sesquiterpenes from the red algae Laurencia obtusa and Laurencia microcladia. Tetrahedron 2006, 62, 182–189.
- Kladi, M.; Vagias, C.; Furnari, G.; Moreau, D.; Roussakis, C.; Roussis, V. Cytotoxic cuparene sesquiterpenes from Laurencia microcladia. Tetrahedron Lett. 2005, 46, 5723–5726.
- Rodrigo, G.; Almanza, G.R.; Cheng, Y.; Peng, J.; Hamann, M.; Duan, R.D.; Åkesson, B. Antiproliferative effects of curcuphenol, a sesquiterpene phenol. Fitoterapia 2010, 81, 762–766.
- Taşdemir, D.; Bugni, T.S.; Mangalindan, G.C.; Concepción, G.P.; Harper, M.K.; Ireland, C.M. Bisabolane type sesquiterpenes from a marine Didiscus sponge. Turkish J. Chem. 2003, 27, 273–279.
- Reddy, P.; Urban, S. Meroditerpenoids from the southern Australian marine brown alga Sargassum fallax. Phytochemistry 2009, 70, 250–255.
- Hur, S.; Lee, H.; Kim, Y.; Lee, B.H.; Shin, J.; Kim, T.Y. Sargaquinoic acid and sargachromenol, extracts of Sargassum sagamianum, induce apoptosis in HaCaT cells and mice skin: Its potentiation of UVB-induced apoptosis. Eur. J. Pharmacol. 2008, 582, 1–11.
- Mori, J.; Iwashima, M.; Wakasugi, H.; Saito, H.; Matsunaga, T.; Ogasawara, M.; Takahashi, S.; Suzuki, H.; Hayashi, T. New plastoquinones isolated from the brown alga, Sargassum micracanthum. Chem. Pharm. Bull. 2005, 53, 1159–1163.
- Bertanha, C.S.; Januário, A.H.; Alvarenga, T.A.; Pimenta, L.P.; E Silva, M.L.A.; Cunha, W.R.; Pauletti, P.M. Quinone and hydroquinone metabolites from the ascidians of the genus Aplidium. Mar. Drugs 2014, 12, 3608–3633.
- García, P.A.; Hernández, Á.P.; San Feliciano, A.; Castro, M.A.Á. Bioactive prenyl- and terpenyl-quinones/ Hydroquinones of marine origin. Mar. Drugs 2018, 16, 292.
- Qu, X.Y.; Ren, J.W.; Peng, A.H.; Lin, S.Q.; Lu, D.D.; Du, Q.Q.; Liu, L.; Li, X.; Li, E.W.; Xie, W.D. Cytotoxic, anti-migration, and anti-invasion activities on breast cancer cells of angucycline glycosides isolated from a marine-derived Streptomyces sp. Mar. Drugs 2019, 17, 277.
- Moon, K.; Chung, B.; Shin, Y.; Rheingold, A.L.; Moore, C.E.; Park, S.J.; Park, S.; Lee, S.K.; Oh, K.B.; Shin, J.; et al. Pentacyclic antibiotics from a tidal mud flat-derived actinomycete. J. Nat. Prod. 2015, 78, 524–529.
- Itoh, T.; Kinoshita, M.; Wei, H.; Kobayashi, M. Stereostructure of komodoquinone A, a neuritogenic anthracycline, from marine Streptomyces sp. KS3. Chem. Pharm. Bull. 2003, 51, 1402–1404.
- Itoh, T.; Kinoshita, M.; Aoki, S.; Kobayashi, M. Komodoquinone A, a Novel Neuritogenic Anthracycline, from Marine Streptomyces sp. KS3. J. Nat. Prod. 2003, 66, 1373–1377.
- Xin, W.; Ye, X.; Yu, S.; Lian, X.Y.; Zhang, Z. New capoamycin-type antibiotics and polyene acids from marine Streptomyces fradiae PTZ0025. Mar. Drugs 2012, 10, 2388–2402.
- Ganesan, S.; Velsamy, G.; Sivasudha, T.; Manoharan, N. MALDI-TOF mass spectrum profiling, antibacterial and anticancer activity of marine Streptomyces fradiae BDMS1. World J. Pharm. Pharm. Sci. 2013, 2, 5148–5165.
- Kita, Y.; Fujioka, H. Marine pyrroloiminoquinone alkaloids. In Topics in Current Chemistry; Springer: Berlin/Heidelberg, Germany, 2012; Volume 309, pp. 131–162. ISBN 9783642255281.
- Kalinski, J.C.J.; Krause, R.W.M.; Parker-Nance, S.; Waterworth, S.C.; Dorrington, R.A. Unlocking the Diversity of Pyrroloiminoquinones Produced by Latrunculid Sponge Species. Mar. Drugs 2021, 19, 68.
- Lin, S.; McCauley, E.P.; Lorig-Roach, N.; Tenney, K.; Naphen, C.N.; Yang, A.M.; Johnson, T.A.; Hernadez, T.; Rattan, R.; Valeriote, F.A.; et al. Another look at pyrroloiminoquinone alkaloids-perspectives on their therapeutic potential from known structures and semisynthetic analogues. Mar. Drugs 2017, 15, 98.
- Dijoux, M.G.; Schnabel, P.C.; Hallock, Y.F.; Boswell, J.L.; Johnson, T.R.; Wilson, J.A.; Ireland, C.M.; Van Soest, R.; Boyd, M.R.; Barrows, L.R.; et al. Antitumor activity and distribution of pyrroloiminoquinones in the sponge genus Zyzzya. Bioorganic Med. Chem. 2005, 13, 6035–6044.
- Radisky, D.C.; Radisky, E.S.; Copp, B.R.; Ireland, C.M.; Barrows, L.R.; Kramer, R.A. Novel Cytotoxic Topoisomerase II Inhibiting Pyrroloiminoquinones from Fijian Sponges of the Genus Zyzzya. J. Am. Chem. Soc. 1993, 115, 1632–1638.
- Goey, A.K.L.; Chau, C.H.; Sissung, T.M.; Cook, K.M.; Venzon, D.J.; Castro, A.; Ransom, T.R.; Henrich, C.J.; McKee, T.C.; McMahon, J.B.; et al. Screening and Biological Effects of Marine Pyrroloiminoquinone Alkaloids: Potential Inhibitors of the HIF-1α/p300 Interaction. J. Nat. Prod. 2016, 79, 1267–1275.
- Aburjania, Z.; Whitt, J.D.; Jang, S.; Nadkarni, D.H.; Chen, H.; Rose, J.B.; Velu, S.E.; Jaskula-Sztul, R. Synthetic Makaluvamine Analogs Decrease c-Kit Expression and Are Cytotoxic to Neuroendocrine Tumor Cells. Molecules 2020, 25, 4940.
- Boucle, S.; Melin, C.; Clastre, M.; Guillard, J. Design, synthesis and evaluation of new marine alkaloid-derived pentacyclic structures with anti-tumoral potency. Mar. Drugs 2015, 13, 655–665.
- Backenköhler, J.; Spindler, S.; Spiteller, P. Total Synthesis of Damirone C, Makaluvamine O, Makaluvone, Batzelline C and Batzelline D. ChemistrySelect 2017, 2, 2589–2592.
- Schneemann, I.; Kajahn, I.; Ohlendorf, B.; Zinecker, H.; Erhard, A.; Nagel, K.; Wiese, J.; Imhoff, J.F. Mayamycin, a cytotoxic polyketide from a Streptomyces strain isolated from the marine sponge Halichondria panicea. J. Nat. Prod. 2010, 73, 1309–1312.
- Liang, Y.; Xie, X.; Chen, L.; Yan, S.; Ye, X.; Anjum, K.; Huang, H.; Lian, X.; Zhang, Z. Bioactive Polycyclic Quinones from Marine Streptomyces sp. 182SMLY. Mar. Drugs 2016, 14, 10–20.
- Wu, K.; Wang, M.; Yao, Q.; Zhang, A. Synthesis study toward mayamycin. Chinese J. Chem. 2013, 31, 93–99.
- Chakraborty, S.; Mal, D. A Representative Synthetic Route for C5 Angucycline Glycosides: Studies Directed toward the Total Synthesis of Mayamycin. J. Org. Chem. 2018, 83, 1328–1339.
- Vicente, J.; Stewart, A.K.; Van Wagoner, R.M.; Elliott, E.; Bourdelais, A.J.; Wright, J.L.C. Monacyclinones, new angucyclinone metabolites isolated from Streptomyces sp. M7-15 associated with the Puerto Rican Sponge Scopalina ruetzleri. Mar. Drugs 2015, 13, 4682–4700.
- Hu, Y.; Martinez, E.D.; MacMillan, J.B. Anthraquinones from a marine-derived Streptomyces spinoverrucosus. J. Nat. Prod. 2012, 75, 1759–1764.
- Sottorff, I.; Künzel, S.; Wiese, J.; Lipfert, M.; Preußke, N.; Sönnichsen, F.D.; Imhoff, J.F. Antitumor anthraquinones from an Easter island sea anemone: Animal or bacterial origin? Mar. Drugs 2019, 17, 154.
- Papendorf, O.; König, G.M.; Wright, A.D. Hierridin B and 2,4-dimethoxy-6-heptadecyl-phenol, secondary metabolites from the cyanobacterium Phormidium ectocarpi with antiplasmodial activity. Phytochemistry 1998, 49, 2383–2386.
- Leão, P.N.; Costa, M.; Ramos, V.; Pereira, A.R.; Fernandes, V.C.; Domingues, V.F.; Gerwick, W.H.; Vasconcelos, V.M.; Martins, R. Antitumor Activity of Hierridin B, a Cyanobacterial Secondary Metabolite Found in both Filamentous and Unicellular Marine Strains. PLoS ONE 2013, 8, e69562.
- Freitas, S.; Martins, R.; Costa, M.; Leão, P.N.; Vitorino, R.; Vasconcelos, V.; Urbatzka, R. Hierridin B Isolated from a Marine Cyanobacterium Alters VDAC1, Mitochondrial Activity, and Cell Cycle Genes on HT-29 Colon Adenocarcinoma Cells. Mar. Drugs 2016, 14, 158.
- Brandão, P.; Moreira, J.; Almeida, J.; Nazareth, N.; Sampaio-Dias, I.E.; Vasconcelos, V.; Martins, R.; Leão, P.; Pinto, M.; Saraíva, L.; et al. Norhierridin B, a new hierridin B-based hydroquinone with improved antiproliferative activity. Molecules 2020, 25, 1578.
- Tian, Y.; Lin, X.; Zhou, X.; Liu, Y. Phenol derivatives from the sponge-derived fungus Didymellaceae sp. SCSIO F46. Front. Chem. 2018, 6, 536–543.
- Li, Z.X.; Wang, X.F.; Ren, G.W.; Yuan, X.L.; Deng, N.; Ji, G.X.; Li, W.; Zhang, P. Prenylated diphenyl ethers from the marine algal-derived endophytic fungus Aspergillus tennesseensis. Molecules 2018, 23, 2368.
- El-Kashef, D.H.; Youssef, F.S.; Reimche, I.; Teusch, N.; Müller, W.E.G.; Lin, W.; Frank, M.; Liu, Z.; Proksch, P. Polyketides from the marine-derived fungus Aspergillus falconensis: In silico and in vitro cytotoxicity studies. Bioorganic Med. Chem. 2021, 29, 115883.
- Liu, Q.Y.; Zhou, T.; Zhao, Y.Y.; Chen, L.; Gong, M.W.; Xia, Q.W.; Ying, M.G.; Zheng, Q.H.; Zhang, Q.Q. Antitumor effects and related mechanisms of Penicitrinine A, a novel alkaloid with a unique spiro skeleton from the marine fungus Penicillium citrinum. Mar. Drugs 2015, 13, 4733–4753.
- Jing, Q.; Hu, X.; Ma, Y.; Mu, J.; Liu, W.; Xu, F.; Li, Z.; Bai, J.; Hua, H.; Li, D. Marine-derived natural lead compound disulfide-linked dimer psammaplin A: Biological activity and structural modification. Mar. Drugs 2019, 17, 384.
- Bao, Y.; Xu, Q.; Wang, L.; Wei, Y.; Hu, B.; Wang, J.; Liu, D.; Zhao, L.; Jing, Y. Studying Histone Deacetylase Inhibition and Apoptosis Induction of Psammaplin A Monomers with Modified Thiol Group. ACS Med. Chem. Lett. 2021, 12, 39–47.
- Ju Han, H.; Sub Byun, W.; Ho Lee, G.; Kyung Kim, W.; Jang, K.; Yang, S.; Yang, J.; Woo Ha, M.; Hong, S.; Lee, J.; et al. Synthesis and biological activity of selenopsammaplin A and its analogues as antitumor agents with DOT1L inhibitory activity. Bioorganic Med. Chem. 2021, 35, 116072.
- Tang, R.; Kimishima, A.; Setiawan, A.; Arai, M. Secalonic acid D as a selective cytotoxic substance on the cancer cells adapted to nutrient starvation. J. Nat. Med. 2020, 74, 495–500.
- Zhang, H.; Huang, L.; Tao, L.; Zhang, J.; Wang, F.; Zhang, X.; Fu, L. Secalonic acid D induces cell apoptosis in both sensitive and ABCG2-overexpressing multidrug resistant cancer cells through upregulating c-Jun expression. Acta Pharm. Sin. B 2019, 9, 516–525.
- Guru, S.K.; Pathania, A.S.; Kumar, S.; Ramesh, D.; Kumar, M.; Rana, S.; Kumar, A.; Malik, F.; Sharma, P.R.; Chandan, B.K.; et al. Secalonic Acid-D represses HIF1α/VEGF-mediated angiogenesis by regulating the Akt/mTOR/p70S6K signaling cascade. Cancer Res. 2015, 75, 2886–2896.
- Sun, H.L.; Zhang, J.; Pan, X.H.; Yan, M.M.; Liu, D.S.; Gao, X.; Zhang, J.R. Secalonic acid- F inhibited cell growth more effectively than 5-fluorouracil on hepatocellular carcinoma in vitro and in vivo. Neoplasma 2017, 64, 344–350.
- Li, N.; Yi, Z.; Wang, Y.; Zhang, Q.; Zhong, T.; Qiu, Y.; Wu, Z.; Tang, X. Differential proteomic analysis of HL60 cells treated with secalonic acid F reveals caspase 3-induced cleavage of Rho GDP dissociation inhibitor 2. Oncol. Rep. 2012, 28, 2016–2022.
- Zeng, Y.; Ren, M.; Li, Y.; Liu, Y.; Chen, C.; Su, J.; Su, B.; Xia, H.; Liu, F.; Jiang, H.; et al. Knockdown of RhoGDI2 represses human gastric cancer cell proliferation, invasion and drug resistance via the Rac1/Pak1/LIMK1 pathway. Cancer Lett. 2020, 492, 136–146.
- Xie, L.; Li, M.; Liu, D.; Wang, X.; Wang, P.; Dai, H.; Yang, W.; Liu, W.; Hu, X.; Zhao, M. Secalonic acid-F, a novel mycotoxin, represses the progression of hepatocellular carcinoma via MARCH1 regulation of the PI3K/AKT/β-catenin signaling pathway. Molecules 2019, 24, 393.
- Özenver, N.; Dawood, M.; Fleischer, E.; Klinger, A.; Efferth, T. Chemometric and transcriptomic profiling, microtubule disruption and cell death induction by secalonic acid in tumor cells. Molecules 2020, 25, 3224.
More