Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Conner Chen and Version 1 by Sen Zhang.
Apoptosis is a normal physiological process of highly regulated cell death that occurs in most multicellular organisms. Apoptosis plays an important role in the cell cycle and is an integral part of the immune system under physiological and pathological conditions. Disorders of apoptosis are associated with autoimmune diseases, bacterial and viral diseases, heart disease, and neurodegeneration. Apoptosis is defined as an energy-dependent cell death which is one of the pathological characteristics of ischemia/reperfusion injury (IRI).
- mitochondria pathway
- apoptosis
- mitochondrial permeability transition pore
- Bcl-2
- nitric oxide
1. Introduction
Apoptosis is a normal physiological process of highly regulated cell death that occurs in most multicellular organisms [1]. Apoptosis plays an important role in the cell cycle and is an integral part of the immune system under physiological and pathological conditions [2]. Disorders of apoptosis are associated with autoimmune diseases, bacterial and viral diseases, heart disease, and neurodegeneration [3]. Apoptosis is defined as an energy-dependent cell death which is one of the pathological characteristics of ischemia/reperfusion injury (IRI) [4]. There are two different pathways of cell apoptosis, the extrinsic death receptor pathway and the intrinsic mitochondrial pathway. Irreversible intracellular genomic damage is caused by various stimuli including gamma-ray irradiation, endoplasmic reticulum stress, growth factor deprivation, and oxidative stress, which activates the intrinsic pathway, known as the mitochondrial pathway [5,6][5][6]. This pathway is responsible for mitochondrial electron transport chain breakage, reactive oxygen species (ROS) production, adenosine triphosphate (ATP) depletion, mitochondrial membrane potential (ΔΨm) decrease, and mitochondrial permeability transition pore (MPTP) opening, thus leading to cell apoptosis [7].
Mitochondria is one of the most pivotal places of energy metabolism and is most sensitive to ischemia and hypoxia. Some observe that mitochondria evolved a unique structure composed of two layers of membrane to preserve some basic functions of organelles [8]. The inner and outer membrane of mitochondria form a unique space within mitochondria, called the intermembrane space [9]. The inner membrane is highly impermeable and does not provide porin, but contains specific transport proteins. The mitochondrial outer membrane envelops the whole mitochondria and contacts the cytoplasm directly [10]. The outer membrane of mitochondria contains several intact proteins, known as porin or voltage-dependent anion-selective channel, which contains a channel permeable to <5000 Dalton molecules to transport macromolecules through mitochondrial membrane transfer proteins [11]. Under the action of various injury factors, the entry of apoptosis-related proteins between mitochondrial bilayer membranes into the cytoplasm begins, eventually leading to cell apoptosis. Ischemia and hypoxia usually firstly damage the structure and function of mitochondria in tissue cells [12]. Mitochondria play a key role in the apoptotic signal transduction pathway during cell apoptosis, which is manifested as electron transfer rupture of the mitochondrial respiratory chain, ROS production, ATP depletion, decreased mitochondrial membrane potential, MPTP opening, and even loss of release of outer membrane proteins, thus leading to cell apoptosis [7]. During liver ischemia reperfusion, mitochondrial structure and function are impaired and induce liver cell apoptosis, which are related to the opening of MPTP, the release of apoptosis-related proteins, the regulation of B lymphocytoma-2 gene (Bcl-2) family proteins, mitochondrial dynamics imbalance, and endoplasmic reticulum (ER) stress [13,14,15][13][14][15].
There are two main pathways of apoptosis: exogenous death receptor pathway and endogenous mitochondrial apoptosis pathway. The external pathway refers to the death receptor pathway, activated by ligands and receptors. A variety of mediators, including TNFa, Fas ligand, TRAIL, and TLIA. The ligand TRAIL is activated by TNFa and other factors, and the pro-apoptotic mediators bind to their respective receptors to catalyze the activation of many Caspase8, which further leads to the activation and pro-apoptotic of Caspase3 [16,17][16][17]. The internal pathway is also known as the mitochondrial pathway. MPTP opens when the mitochondrial structure is damaged by external stimuli such as hypoxia, radiation, and cytotoxin. With the pro-apoptotic factors into the cytosol, the Bcl-2 family activates proapoptotic factors [2,18][2][18]. The translocation of Bax to the outer membrane of mitochondria causes changes in mitochondrial membrane permeability, then the mitochondrial transmembrane potential is to reduce and depolarize, releasing cytochrome-C and other active factors in the mitochondrial matrix. Cytochrome-C enters into the cytoplasm and binds to Apaf-1 to form the oligomer under the synergistic effect of ATP/dATP, which activates caspase-9 and downstream caspase-3, leading to cell apoptosis [19].
2. The Initiation of Apoptosis: Mitochondrial Fission and Fusion
Mitochondria keeps its dynamic renewal by its continuous fission and fusion [20]. Mitochondrial fission causes its division, while its fusion leads to the binding and prolongation of phospholipid membranes in mitochondria (Figure 1). Mitochondria are strictly controlled by mitotic proteins embedded in the outer and inner membranes through stimulating mitochondrial fusion and fission. However, the dynamic cycle of fission and fusion in mitochondria is destroyed when under stress or damaged. The damaged dynamics in mitochondria may eventually lead to apoptosis by the excessive fission and reduced fusion in mitochondria after hepatic IRI [21].
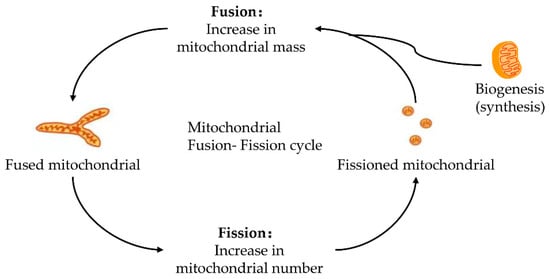
Figure 1. Fusion–fission cycle in mitochondria. Mitochondria strictly control the fusion and fission process through mitotic proteins embedded in the inner and outer membranes to maintain the dynamic balance of biological energy. The mitofusins and Opa1 mediate the fusion of the mitochondrial membrane. Fusion and fission belong to the mitochondrial quality control cycle. The growth and division of pre-existing mitochondria are also involved.
2.1. Mitochondrial Fission and Fission
Mitochondrial fission is due to the increased mitochondrial division or decreased mitochondrial fusion [22,23,24][22][23][24]. The mitochondrial division damaged organelles are responsible for mitochondrial fission and mitosis. These fragmented organelles fuse to an interconnected network which renews the damaged mitochondrial DNA (mtDNA) [25]. Excessive fission causes mitochondrial breakage and activates its apoptosis pathway, thus aggravating tissue damage and cell apoptosis [26]. Additionally, excessive fission of mitochondria is a pre-symptom of cytochrome-C (cyt c) release [27], and the release of cyt c further promotes the mitosis of mitochondria.
Long et al. [21] showed that mitochondrial dynamics are related to the regulation of dynamin-related protein1(Drp1) and Mitochondrial fission protein 1 (MTFP1). Drp1 is a key protein in mitochondrial fission, which is mainly present in the cytoplasm and transferred to mitochondria after activation [28]. Hepatic IRI affects mitochondrial dynamics by translocating Drp1 to mitochondria in a fission-based manner [28]. During mitochondrial fission, the cytoplasm-localized Drp1 was recruited to its outer membrane which mediated membrane division. It is reported that interrupted mitochondrial fission by Drp1 protects hepatocytes from IRI-induced apoptosis [29]. In addition, Drp1-mediated mitochondrial fragmentation is regulated by Drp1 phosphorylation at Ser616 and Ser637, which results in the activation and inactivation of Drp1, respectively [28]. Drp1 phosphorylation at Ser616 promotes mitochondrial fission, and Drp1 phosphorylation at Ser637 seems to induce cell apoptosis. Drp1 Phosphorylation at Ser637 by protein kinase A (PKA) lengthens mitochondria to inhibit cell apoptosis, whereas Drp1 dephosphorylation at Ser637 by calcineurin (CaN) promotes mitochondrial fragmentation [30].
2.2. Mitochondrial Fusion
Mitochondrial fusion has long been regarded as a protective way to reduce mitochondrial fission [9]. During hepatic IRI, the fusion of damaged mitochondria could cause a beneficial effect to maintain the survival function of mitochondria [26] by preventing mitochondrial decomposition caused by the release of cyt c, xanthine oxidase (XO), and ROS in mitochondrial [31]. Fusion is associated with the redistribution of metabolites, proteins, and mtDNA in mitochondria. Fusion is also helpful for maintaining oxidative phosphorylation and integrity of mtDNA, and enhancing the synthesis of ATP [32].
The consequence of fusion is related to intimal proteins optic atrophy 1 (Opa1), mitochondrial fusion protein 1 (Mfn1), and Mfn2. Previous research has established that Opa1-induced fusion may be affected by extracellular regulated protein kinases (ERK) and sirtuin-3 (SIRT3) [33]. The highlight is that SIRT3 maintains mitochondrial homeostasis on IRI by enhancing mitochondrial fusion triggered by Opa1 [32]. Inhibition of ERK eliminates the regulatory effect of SIRT3 on Opa1 expression and mitochondrial fusion, resulting in mitochondrial damage and apoptosis of renal tubular epithelial cells [10].
3. “Switch” Role of Mitochondrial Permeability Transition Pore
MPTP is a group of protein complexes with non-specificity and voltage dependencies present between the inner and outer mitochondrial membranes (Figure 2). The outer membrane of mitochondria contains many intact proteins known as porin, which contains a channel that is permeable to <5000 Dalton molecules to transport macromolecules through mitochondrial membrane transfer proteins [11]. During mitochondrial damage, the inner membrane of the mitochondrial collapses once the mitochondrial permeability transition is initiated, allowing apoptosis-related proteins to rush out of the mitochondrial, and eventually leading to cell apoptosis [34,35][34][35].
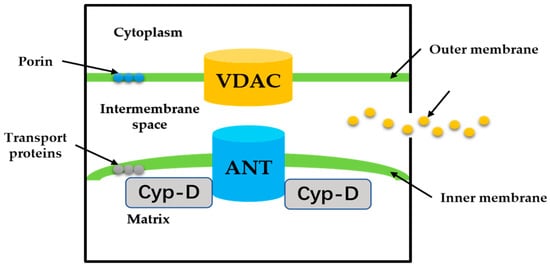
Figure 2. The proposed structure of the MPTP. Cyp-D is a small molecule in the mitochondrial matrix. VDAC on the OMM, ANT on the IMM.
The historical model of the mPTP comprised three components: voltage-dependent anion channel (VDAC) in the outer membrane of mitochondria, adenine nucleotide translocase (ANT) in the inner membrane of mitochondria, and cyclophilin D (CypD) in the mitochondrial matrix. Crompton et al. [36] demonstrated that CsA was an effective inhibitor of MPTP opening, and later studies showed that CsA inhibited pore opening through inhibition of matrix peptidyl-prolyl cis-trans isomerase (PPIase). CsA is an inhibitor of CypD, which proves that the protein on MPTP is CypD [37]. Matrix PPIase activity promotes conformational changes in intimal proteins, but the composition of CypD is still under investigation. Studies have shown that liver mitochondria of CypD knockout mice are highly resistant to calcium-induced MPTP opening [38]. The mitochondrial phosphate carrier (PiC) is consistent with the calcium-induced conformational change of PiC in mPTP formation. Co-immunoprecipitation and GST-CyP-D pull-down analysis showed that CYP-D interacts with PiC [39]. The bovine ANT1 of CAT complex is consistent with ANT, which forms MPTP. It also suggests that ANT has a conformation of a large cavity extending from the cytoplasmic side into the membrane, in which a contraction channel comprising three spirals prevents pore formation [40,41][40][41]. If conformational changes facilitated by CYP-D rearranged these helices, this may also be responsible for the formation of MPTP.
As a dynamic structure that closes during ischemia and opens during reperfusion, MPTP plays a “switch” role in apoptosis [42,43,44]. MPTP brings destruction to the proton gradient and electric potential of the mitochondrial inner membrane, leads to the inflow of solutes and water, and increases the permeability of mitochondria, thus increasing the burden on the mitochondrial. Subsequent swelling and breakdown of the outer membrane activate the cascade reaction of pro-apoptosis [45,46,47,48]. It can thus be suggested that pro-apoptotic proteins are released into the intracellular mediator, which contributes to the release of Cyt c, apoptosis inducing factor (AIF), and the formation of apoptotic bodies [8]. Another important finding was that ATP levels decreased significantly when most hepatocyte mitochondria were affected by MPTP [49]. Recent cases reported by Sun et al. [50] also support the hypothesis that this decline in ATP levels is often accompanied by mitochondria swelling, activation of caspases, up-regulation of Bcl-2, and downregulation of Bcl-2-associated X protein (Bax). Even if a small fraction of MPTP lifts a restriction, mitochondrial homeostasis imbalance and cell apoptosis will still occur [20,51]. Surveys conducted by Cai et al. [52] have shown that the cell apoptosis was effectively offset by inhibition of MPTP opening. During hepatic IRI, MPTP opening is induced by the following alterations: over-production of ROS, calcium (CaAs a dynamic structure that closes during ischemia and opens during reperfusion, MPTP plays a “switch” role in apoptosis [42][43][44]. MPTP brings destruction to the proton gradient and electric potential of the mitochondrial inner membrane, leads to the inflow of solutes and water, and increases the permeability of mitochondria, thus increasing the burden on the mitochondrial. Subsequent swelling and breakdown of the outer membrane activate the cascade reaction of pro-apoptosis [45][46][47][48]. It can thus be suggested that pro-apoptotic proteins are released into the intracellular mediator, which contributes to the release of Cyt c, apoptosis inducing factor (AIF), and the formation of apoptotic bodies [8]. Another important finding was that ATP levels decreased significantly when most hepatocyte mitochondria were affected by MPTP [49]. Recent cases reported by Sun et al. [50] also support the hypothesis that this decline in ATP levels is often accompanied by mitochondria swelling, activation of caspases, up-regulation of Bcl-2, and downregulation of Bcl-2-associated X protein (Bax). Even if a small fraction of MPTP lifts a restriction, mitochondrial homeostasis imbalance and cell apoptosis will still occur [20][51]. Surveys conducted by Cai et al. [52] have shown that the cell apoptosis was effectively offset by inhibition of MPTP opening. During hepatic IRI, MPTP opening is induced by the following alterations: over-production of ROS, calcium (Ca2+) overload, and ∆Ψm loss (Table 1).
Table 1. Influencing factors of MPTP opening. Excessive ROS, elevated calcium, and decreased ΔΨm; these factors are interconnected and can lead to the same events: the MPT pore opening, damage to mitochondrial membranes, and release of pro-apoptotic proteins.
| Factors | How | Effects | Results |
|---|---|---|---|
| ROS | excessive ROS produce after reperfusion and CO exposure | promote mitochondrial permeability transition and depolarizes ΔΨm; produce lipid peroxides and other toxic aldehydes | induce MPTP opening |
| Ca2+ | Na/CaNa/Ca+2+ commutator overburden leads to Ca2+ commutator overburden leads to Ca2+ overload overload | induce PKC formation activate NFkB activate Ca2+-dependent enzymes; cause mitochondrial integrity impairment | mitochondrial membrane damage |
| ΔΨm | mitochondrial integrity impaired causes the ΔΨm loss | block the synthesis of mitochondrial RNA and protein, uncoupling oxidative phosphorylation, and ATP depletion | release of apoptosis drivers, cyt c |
3.1. ROS Triggered MPTP Opening during Hepatic IRI
The early characteristics of hepatic IRI are the occurrence of oxidative stress and the release of ROS, which directly lead to hepatocyte injury [53]. Mitochondria are both targets and generators of ROS [54]. Under normal physiological states, hepatocytes can withstand certain levels of ROS. However, excessive ROS can be produced when damaged mitochondria become dysfunctional, resulting in greater damage to mitochondria [55,56][55][56]. Thus far, previous studies have suggested that excessive ROS production during hepatic IRI can result in apoptosis due to the further damage of DNA, proteins, lipids, and other cellular molecules [57].
Evidence suggests that the overproduction of ROS mainly occurs after blood and oxygen return to the hypoxic liver [58]. This increase in ROS allows for mitochondrial permeability transition, depolarization of ΔΨm, and MPTP opening [59]. Another significant aspect of ROS-induced MPTP opening is that ROS interacts with polyunsaturated fatty acids in biofilms to produce highly toxic and reactive molecules, resulting in the production of lipid peroxides and poisonous aldehydes, such as 4-hydroxynonenal (4-HNE) or malondialdehyde (MDA). Firstly, the accumulation of 4-HNE may activate a series of signaling pathways that lead to liver cell apoptosis, including decreased integrity of the mitochondrial membrane, increased permeability of mitochondrial, and inhibited electron transport chain [60,61][60][61]. Secondly, MDA is a major metabolite of lipid peroxides that have been demonstrated to be the main cause of cell membrane damage [62].
As key binding targets for carbon monoxide (CO), mitochondria are an important target for CO-dependent regulation of cellular physiology and signal pathways due to the richness in iron such as heme [63]. The toxicity of CO on mitochondria has been broadened to relate to ROS generation, ΔΨm, mitochondrial respiration, and mitochondrial-dependent metabolic pathways. As noted by Jung et al. [64], CO regulates the production of mitochondrial ROS in a specific manner. At high concentrations, CO can inhibit mitochondrial respiration and ATP production, and regulate the glycolytic pathway in a dose-dependent manner [65]. On the other hand, as the key catalyst for the formation of ROS, iron is the main instigator of MPTP. Intracellular chelated iron can promote hepatocyte oxidative damage and MPTP-induced apoptosis [66]. During the phase of ischemic, the lysosome releases chelatable ferrous iron (Fe2+), and then Fe2+ is absorbed into mitochondria by mitochondrial Ca2+ uniporter. Once the Fe2+ overload occurs in mitochondria, the formation of hydrogen peroxide after reperfusion leads to the production of hydroxyl radicals (OHD). In particular, OHD will damage DNA, protein, and cell membranes, resulting in MPTP opening and cell apoptosis [67].
3.2. Calcium Overload and MPTP Opening during IRI
The imbalance of Ca2+ homeostasis is a common way that has a considerable impact on hepatocyte injury. Intracellular Ca2+ concentration is approximately 10–100 nM, 10,000 times lower than extracellular Ca2+ concentration. This gradient can be maintained by four mechanisms: (1) ATP-mediated transmembrane outflow; (2) Na/K-mediated Ca++2+/Na retrograde transport; (3) Ca+2+ storage capacity of endoplasmic reticulum; and (4) oxygen-dependent intracellular Ca2+ pump in mitochondria [68,69]. However, with the increased concentration of cytoplasmic Ca pump in mitochondria [68][69]. However, with the increased concentration of cytoplasmic Ca2+, mitochondria can act as a buffer for redundant Ca2+. This subsequently leads to the migration of abundant intracellular Ca2+ due to the increase in intracellular Na concentration and the antiport of Na/Ca++2+. Moreover, ischemia and hypoxia lead to an increase in cell membrane permeability, resulting in a further pile-up of the intracellular Ca2+. Ischemia and hypoxia also violate the structure and function of mitochondria, resulting in the release of large amounts of Ca2+ from the endoplasmic reticulum (ER) and intracellular Ca2 overload [70].+
With the development of research, the molecular structure of mitochondrial calcium ion transport has been identified as mitochondrial Ca2+ uniporter (MCU), Na/Ca+2+ exchanger (NCLX) and Ca2+/H antiporter (Letm1) [71]. The consensus is that MCU is primarily responsible for mitochondrial Ca+2+ influx and that MCU promotes Ca2+ transport down its electrochemical gradient. MICU1 is an adjustable MCU containing Ca2+ binding EF-Hand structure [72]. When intracellular Ca2+ concentration is low, it prevents Ca2+ from entering the MCU channel. The MiCU1/MiCU2 ratio, and its interaction with MCU, determine the dynamics of Ca2+ transport to mitochondria. Overexpression of MICU1 gene also results in significantly sped up Ca2+ entry into mitochondria [72]. Letm1(K/H exchanger), mediates mitochondrial Ca++2+ and H transport. With the dual role of Letm1 as a Ca+2+/H exchanger, Letm1 transports Ca+2+ in and out of mitochondria in a Ca2+ and pH gradient-dependent manner [72]. When I/R injury occurs, the level of intracellular ATP decreases, resulting in down-regulation of the activity of ATP-dependent Na/K-AT-pase embedded in the cell membrane [73].++
Mitochondria Ca2+ overload can promote lipid peroxidation and weaken the oxidative phosphorylation of mitochondria, resulting in impaired structure and function of mitochondria and decreased ATP synthesis rate [69]. Interestingly, the damaged mitochondrial membrane structure caused by lipid peroxidation aggravates Ca2+ overload. Therefore, both mitochondria Ca2+ overload and lipid peroxidation generally occur in liver damage after IRI. Further analysis showed that the increase in intracellular Ca2+ during the ischemia period also promoted the production of XO. Chang et al. [74] conclude that cell reoxygenation in the process of reperfusion causes XO to induce the production of superoxide and the restoration of ATP levels, which allows mitochondria to actively uptake Ca2+, resulting in a large amount of Ca2+ overload. To maintain the integrity of the mitochondrial membrane, the mitochondrial ATP synthase reverses its activity to provide energy for different ion pumps in the mitochondrial membrane [75]. However, this further increases the inflow of Ca2+. Mitochondrial Ca2+ overload leads to mitochondrial membrane damage, especially the decrease in mitochondrial transmembrane potential and MPTP opening. As a result, pro-apoptotic factors are released to the cytoplasm and accelerate cell death [76].
The relationship between Ca2+ and MPTP opening is complex and related to a variety of different pathways. Ca2+-induced MPTP opening has three separable but interrelated mechanisms. Firstly, hypoxia has been reported to be an effective disruptor of oxidative phosphorylation, hindering the production of ATP and leading to Ca2+ overload [20]. It was suggested that the extent mitochondrial Na/Ca+2+ commutator overburdened and inactive state exacerbated with Ca2+ overload, causing the concentration of Ca2+ to increase enough to trigger the activation of MPTP opening [26,74]. Secondly, the increase in intracellular Ca to increase enough to trigger the activation of MPTP opening [26][74]. Secondly, the increase in intracellular Ca2+ content induces the formation of protein kinase C (PKC). Nakazato et al. [77] points out that PKC shows a strong indirect elevating effect on nuclear transcription factor kappa B (NFkB) activation and ROS production, thus promoting MPTP opening. Lastly, intracellular Ca2+ overload can activate Ca2+-dependent enzymes, such as calpain, and phospholipase C [78]. Calpain is a Ca2+-dependent intracellular cysteine protease [79]. An uncontrolled increase in Ca2+ levels can lead to continuous activation of calpain. Cannistra et al. [66] suggested that calpain-induced degradation of autophagy-related 7 and Beclin-1 leads to autophagy defect and MPTP-dependent hepatocyte death after IRI.
3.3. Mitochondrial Membrane Potential Loss and MPTP Opening during IRI
ΔΨm is an increasingly important aspect in maintaining mitochondrial function and inhibiting hepatocyte apoptosis [80]. The loss of ΔΨm is one of the earliest events in the cascade of apoptosis. Under normal physiological conditions, the existence of ΔΨm mainly depends on the closed MPTP [5]. In light of recent research, MPTP opening destroys the integrity of the mitochondrial membrane and makes the ΔΨm lose or collapse [74]. After ΔΨm loss, the synthesis of mtRNA and protein was blocked, followed by uncoupling oxidative phosphorylation and ATP depletion, resulting in osmotic swelling and outer membrane rupture, leading to mitochondrial permeability transition, releasing apoptosis-driving factors such as cyt c, which leads to cell apoptosis [81].
Ca2+/calmodulin dependent protein kinase II (CaMKII) proved an important member of the CaMK family. CaMKII is a kind of protein activated by Ca2+ and calmodulin. Zhang et al. [7] demonstrated that CaMKIIγ could induce the change of ΔΨm and mitochondrial permeability. In the same vein, Kang et al. [82] found that the overexpression of CaMKII γ caused significant ultrastructural damage, such as mitochondria swelling, hepatocyte necrosis, mitochondria membrane rupture, and atrophy. Furthermore, phospholipases of CaMKII can regulate the influx of Ca2+ into mitochondria [76]. This mechanism is similar to that reported by Joiner et al. [83] who report CaMK II activity can regulate the influx of Ca2+ into mitochondria and promote apoptosis.
3.4. Regulatory Role of Akt/GSK-3β Pathway on MPTP Opening
The Akt/GSK-3β pathway has significant biological functions in MPTP opening. Many prosurvival signaling pathways inactivate glycogen GSK-3β by regulating phosphorylated GSK-3β, then increase the opening of MPTP and regulate the IRI [84]. The reperfusion injury salvage kinase (RISK) and survivor activating factor enhancement (SAFE) pathways are considered the two main pathways of MPTP opening [85,86][85][86]. To date, previous research has shown that GSK-3β at Ser9 is phosphorylated by Akt, resulting in the interaction with MPTP regulatory factors and inhibiting the opening of MPTP during reperfusion [45].
At the same time, the phosphorylation of GSK-3β can also actively regulate β-Catenin [87]. Zhao et al. [84] demonstrate that administration of GSK-3β inhibitors before IRI can increase the accumulation of intracellular β-catenin, thus activating the GSK-3β/β-catenin signaling pathway and further enhancing the expression of Bcl-2.
References
- Kerr, J.F. History of the events leading to the formulation of the apoptosis concept. Toxicology 2002, 181, 471–474.
- Xu, X.; Lai, Y.; Hua, Z.C. Apoptosis and apoptotic body: Disease message and therapeutic target potentials. Biosci. Rep. 2019, 39, BSR20180992.
- Wang, M.L.; Tuli, R.; Manner, P.A.; Sharkey, P.F.; Hall, D.J.; Tuan, R.S. Direct and indirect induction of apoptosis in human mesenchymal stem cells in response to titanium particles. J. Orthop. Res. Off. Publ. Orthop. Res. Soc. 2003, 21, 697–707.
- Bi, J.; Zhang, J.; Ren, Y.; Du, Z.; Li, Q.; Wang, Y.; Wei, S.; Yang, L.; Zhang, J.; Liu, C.; et al. Irisin alleviates liver ischemia-reperfusion injury by inhibiting excessive mitochondrial fission, promoting mitochondrial biogenesis and decreasing oxidative stress. Redox Biol. 2019, 20, 296–306.
- Chu, W.W.; He, X.Y.; Yan, A.L.; Wang, S.W.; Li, S.; Nian, S.; Wang, Y.L.; Liang, F.L. Ischemic postconditioning lightening ischemia/reperfusion apoptosis of rats via mitochondria pathway. Eur. Rev. Med. Pharm. Sci. 2019, 23, 6307–6314.
- Guicciardi, M.E.; Malhi, H.; Mott, J.L.; Gores, G.J. Apoptosis and Necrosis in the Liver. Compr. Physiol. 2013, 3, 977–1010.
- Zhang, L.; Zhang, S.N.; Li, L.; Zhang, X.B.; Wu, R.C.; Liu, J.H. Prolonged warm ischemia aggravates hepatic mitochondria damage and apoptosis in DCD liver by regulating Ca(2+)/CaM/CaMKII signaling pathway. Int. J. Clin. Exp. Pathol. 2019, 12, 217–228.
- Serviddio, G.; Bellanti, F.; Sastre, J.; Vendemiale, G.; Altomare, E. Targeting mitochondria: A new promising approach for the treatment of liver diseases. Curr. Med. Chem. 2010, 17, 2325–2337.
- Jiang, S.J.; Li, W.; An, W. Adenoviral gene transfer of hepatic stimulator substance confers resistance against hepatic ischemia-reperfusion injury by improving mitochondrial function. Hum. Gene Ther. 2013, 24, 443–456.
- Guan, L.Y.; Fu, P.Y.; Li, P.D.; Li, Z.N.; Liu, H.Y.; Xin, M.G.; Li, W. Mechanisms of hepatic ischemia-reperfusion injury and protective effects of nitric oxide. World J. Gastrointest. Surg. 2014, 6, 122–128.
- Zhang, X.; Lemasters, J.J. Translocation of iron from lysosomes to mitochondria during ischemia predisposes to injury after reperfusion in rat hepatocytes. Free Radic. Biol. Med. 2013, 63, 243–253.
- Brekke, E.; Berger, H.R.; Wideroe, M.; Sonnewald, U.; Morken, T.S. Glucose and Intermediary Metabolism and Astrocyte-Neuron Interactions Following Neonatal Hypoxia-Ischemia in Rat. Neurochem. Res. 2017, 42, 115–132.
- Zhang, H.; Yan, Q.; Wang, X.; Chen, X.; Chen, Y.; Du, J.; Chen, L. The Role of Mitochondria in Liver Ischemia-Reperfusion Injury: From Aspects of Mitochondrial Oxidative Stress, Mitochondrial Fission, Mitochondrial Membrane Permeable Transport Pore Formation, Mitophagy, and Mitochondria-Related Protective Measures. Oxid. Med. Cell. Longev. 2021, 2021, 6670579.
- Masior, L.; Grat, M. Methods of Attenuating Ischemia-Reperfusion Injury in Liver Transplantation for Hepatocellular Carcinoma. Int. J. Mol. Sci. 2021, 22, 8229.
- Zhang, Q.; Liu, X.; Piao, C.; Jiao, Z.; Ma, Y.; Wang, Y.; Liu, T.; Xu, J.; Wang, H. Effect of conditioned medium from adipose derived mesenchymal stem cells on endoplasmic reticulum stress and lipid metabolism after hepatic ischemia reperfusion injury and hepatectomy in swine. Life Sci. 2022, 289, 120212.
- Qiu, F.; Hu, M.; Tang, B.; Liu, X.; Zhuang, H.; Yang, J.; Hua, Z.C. Annexin V-TRAIL fusion protein is a more sensitive and potent apoptotic inducer for cancer therapy. Sci. Rep. 2013, 3, 3565.
- Wu, M.Y.; Yiang, G.T.; Liao, W.T.; Tsai, A.P.; Cheng, Y.L.; Cheng, P.W.; Li, C.Y.; Li, C.J. Current Mechanistic Concepts in Ischemia and Reperfusion Injury. Cell Physiol. Biochem. 2018, 46, 1650–1667.
- Batandier, C.; Leverve, X.; Fontaine, E. Opening of the mitochondrial permeability transition pore induces reactive oxygen species production at the level of the respiratory chain complex I. J. Biol. Chem. 2004, 279, 17197–17204.
- Wu, Q.; Tang, C.; Zhang, Y.J.; Jiang, Y.; Li, X.W.; Wang, S.G.; Bie, P. Diazoxide suppresses hepatic ischemia/reperfusion injury after mouse liver transplantation by a BCL-2-dependent mechanism. J. Surg. Res. 2011, 169, e155–e166.
- Rossetti, A.; Togliatto, G.; Rolo, A.P.; Teodoro, J.S.; Granata, R.; Ghigo, E.; Columbano, A.; Palmeira, C.M.; Brizzi, M.F. Unacylated ghrelin prevents mitochondrial dysfunction in a model of ischemia/reperfusion liver injury. Cell Death Discov. 2017, 3, 17077.
- Long, R.T.; Peng, J.B.; Huang, L.L.; Jiang, G.P.; Liao, Y.J.; Sun, H.; Hu, Y.D.; Liao, X.H. Augmenter of Liver Regeneration Alleviates Renal Hypoxia-Reoxygenation Injury by Regulating Mitochondrial Dynamics in Renal Tubular Epithelial Cells. Mol. Cells 2019, 42, 893–905.
- Andrieux, P.; Chevillard, C.; Cunha-Neto, E.; Nunes, J.P.S. Mitochondria as a Cellular Hub in Infection and Inflammation. Int. J. Mol. Sci. 2021, 22, 1338.
- Lechado Terradas, A.; Zittlau, K.I.; Macek, B.; Fraiberg, M.; Elazar, Z.; Kahle, P.J. Regulation of mitochondrial cargo-selective autophagy by posttranslational modifications. J. Biol. Chem. 2021, 297, 101339.
- Ding, Q.; Qi, Y.; Tsang, S.Y. Mitochondrial Biogenesis, Mitochondrial Dynamics, and Mitophagy in the Maturation of Cardiomyocytes. Cells 2021, 10, 2463.
- Wai, T.; Langer, T. Mitochondrial Dynamics and Metabolic Regulation. Trends Endocrinol. Metab. 2016, 27, 105–117.
- Gu, J.; Zhang, T.; Guo, J.; Chen, K.; Wang, G.; Li, H.; Wang, J. Ursodeoxycholyl lysophosphatidylethanolamide protects against hepatic ischemia/reperfusion injury via phospholipid metabolism-mediated mitochondrial quality control. FASEB J. 2020, 34, 6198–6214.
- Tilokani, L.; Nagashima, S.; Paupe, V.; Prudent, J. Mitochondrial dynamics: Overview of molecular mechanisms. Essays Biochem. 2018, 62, 341–360.
- Yu, X.; Jia, L.; Yu, W.; Du, H. Dephosphorylation by calcineurin regulates translocation of dynamin-related protein 1 to mitochondria in hepatic ischemia reperfusion induced hippocampus injury in young mice. Brain Res. 2019, 1711, 68–76.
- Zhang, C.; Huang, J.; An, W. Hepatic stimulator substance resists hepatic ischemia/reperfusion injury by regulating Drp1 translocation and activation. Hepatology 2017, 66, 1989–2001.
- Kraus, F.; Roy, K.; Pucadyil, T.J.; Ryan, M.T. Function and regulation of the divisome for mitochondrial fission. Nature 2021, 590, 57–66.
- Qajari, N.M.; Shafaroudi, M.M.; Gholami, M.; Khonakdar-Tarsi, A. Silibinin treatment results in reducing OPA1&MFN1 genes expression in a rat model hepatic ischemia–reperfusion. Mol. Biol. Rep. 2020, 47, 3271–3280.
- Wang, Q.; Xu, J.; Li, X.; Liu, Z.; Han, Y.; Xu, X.; Li, X.; Tang, Y.; Liu, Y.; Yu, T.; et al. Sirt3 modulate renal ischemia-reperfusion injury through enhancing mitochondrial fusion and activating the ERK-OPA1 signaling pathway. J. Cell. Physiol. 2019, 234, 23495–23506.
- Jaeschke, H.; Woolbright, B.L. Current strategies to minimize hepatic ischemia-reperfusion injury by targeting reactive oxygen species. Transpl. Rev. 2012, 26, 103–114.
- Martins, R.M.; Teodoro, J.S.; Furtado, E.; Rolo, A.P.; Palmeira, C.M.; Tralhao, J.G. Recent insights into mitochondrial targeting strategies in liver transplantation. Int. J. Med. Sci. 2018, 15, 248–256.
- Sullivan, E.M.; Pennington, E.R.; Green, W.D.; Beck, M.A.; Brown, D.A.; Shaikh, S.R. Mechanisms by Which Dietary Fatty Acids Regulate Mitochondrial Structure-Function in Health and Disease. Adv. Nutr. 2018, 9, 247–262.
- Crompton, M.; Ellinger, H.; Costi, A. Inhibition by cyclosporin A of a Ca2+-dependent pore in heart mitochondria activated by inorganic phosphate and oxidative stress. Biochem. J. 1988, 255, 357–360.
- Baines, C.P.; Kaiser, R.A.; Purcell, N.H.; Blair, N.S.; Osinska, H.; Hambleton, M.A.; Brunskill, E.W.; Sayen, M.R.; Gottlieb, R.A.; Dorn, G.W.; et al. Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature 2005, 434, 658–662.
- Basso, E.; Fante, L.; Fowlkes, J.; Petronilli, V.; Forte, M.A.; Bernardi, P. Properties of the permeability transition pore in mitochondria devoid of Cyclophilin D. J. Biol. Chem. 2005, 280, 18558–18561.
- Leung, A.W.; Varanyuwatana, P.; Halestrap, A.P. The mitochondrial phosphate carrier interacts with cyclophilin D and may play a key role in the permeability transition. J. Biol. Chem. 2008, 283, 26312–26323.
- Marzo, I.; Brenner, C.; Zamzami, N.; Susin, S.A.; Beutner, G.; Brdiczka, D.; Rémy, R.; Xie, Z.H.; Reed, J.C.; Kroemer, G. The permeability transition pore complex: A target for apoptosis regulation by caspases and bcl-2-related proteins. J. Exp. Med. 1998, 187, 1261–1271.
- Pebay-Peyroula, E.; Dahout-Gonzalez, C.; Kahn, R.; Trézéguet, V.; Lauquin, G.J.; Brandolin, G. Structure of mitochondrial ADP/ATP carrier in complex with carboxyatractyloside. Nature 2003, 426, 39–44.
- Wallimann, T.; Riek, U.; Moddel, M. Intradialytic creatine supplementation: A scientific rationale for improving the health and quality of life of dialysis patients. Med. Hypotheses 2017, 99, 1–14.
- Zaouali, M.A.; Panisello, A.; Lopez, A.; Castro, C.; Folch, E.; Carbonell, T.; Rolo, A.; Palmeira, C.M.; Garcia-Gil, A.; Adam, R.; et al. GSK3β and VDAC Involvement in ER Stress and Apoptosis Modulation during Orthotopic Liver Transplantation. Int. J. Mol. Sci. 2017, 18, 591.
- Khan, H.A.; Ahmad, M.Z.; Khan, J.A.; Arshad, M.I. Crosstalk of liver immune cells and cell death mechanisms in different murine models of liver injury and its clinical relevance. Hepatobiliary Pancreat. Dis. Int. 2017, 16, 245–256.
- Zhang, Q.; Fu, H.; Zhang, H.; Xu, F.; Zou, Z.; Liu, M.; Wang, Q.; Miao, M.; Shi, X. Hydrogen sulfide preconditioning protects rat liver against ischemia/reperfusion injury by activating Akt-GSK-3beta signaling and inhibiting mitochondrial permeability transition. PLoS ONE 2013, 8, e74422.
- Brown-Suedel, A.N.; Bouchier-Hayes, L. Caspase-2 Substrates: To Apoptosis, Cell Cycle Control, and Beyond. Front Cell Dev. Biol. 2020, 8, 610022.
- Kroemer, G.; Galluzzi, L.; Brenner, C. Mitochondrial membrane permeabilization in cell death. Physiol. Rev. 2007, 87, 99–163.
- Lin, H.-C.; Lee, T.-K.; Tsai, C.-C.; Lai, I.R.; Lu, K.-S. Ischemic Postconditioning Protects Liver From Ischemia-Reperfusion Injury by Modulating Mitochondrial Permeability Transition. Transplantation 2012, 93, 265–271.
- Weigand, K.; Brost, S.; Steinebrunner, N.; Büchler, M.; Schemmer, P.; Müller, M. Ischemia/Reperfusion Injury in Liver Surgery and Transplantation: Pathophysiology. HPB Surg. 2012, 2012, 1–8.
- Sun, Z.; Lan, X.; Ahsan, A.; Xi, Y.; Liu, S.; Zhang, Z.; Chu, P.; Song, Y.; Piao, F.; Peng, J.; et al. Phosphocreatine protects against LPS-induced human umbilical vein endothelial cell apoptosis by regulating mitochondrial oxidative phosphorylation. Apoptosis 2016, 21, 283–297.
- Lin, J.; Huang, H.F.; Yang, S.K.; Duan, J.; Qu, S.M.; Yuan, B.; Zeng, Z. The effect of Ginsenoside Rg1 in hepatic ischemia reperfusion (I/R) injury ameliorates ischemia-reperfusion-induced liver injury by inhibiting apoptosis. Biomed. Pharm. 2020, 129, 110398.
- Cai, L.; Li, Y.; Zhang, Q.; Sun, H.; Yan, X.; Hua, T.; Zhu, Q.; Xu, H.; Fu, H. Salidroside protects rat liver against ischemia/reperfusion injury by regulating the GSK-3beta/Nrf2-dependent antioxidant response and mitochondrial permeability transition. Eur. J. Pharm. 2017, 806, 32–42.
- Li, H.; Sun, J.J.; Chen, G.Y.; Wang, W.W.; Xie, Z.T.; Tang, G.F.; Wei, S.D. Carnosic acid nanoparticles suppress liver ischemia/reperfusion injury by inhibition of ROS, Caspases and NF-kappaB signaling pathway in mice. Biomed. Pharm. 2016, 82, 237–246.
- Bellanti, F.; Mirabella, L.; Mitarotonda, D.; Blonda, M.; Tamborra, R.; Cinnella, G.; Fersini, A.; Ambrosi, A.; Dambrosio, M.; Vendemiale, G.; et al. Propofol but not sevoflurane prevents mitochondrial dysfunction and oxidative stress by limiting HIF-1alpha activation in hepatic ischemia/reperfusion injury. Free Radic. Biol. Med. 2016, 96, 323–333.
- Schlegel, A.; Muller, X.; Dutkowski, P. Hypothermic Machine Preservation of the Liver: State of the Art. Curr. Transplant. Rep. 2018, 5, 93–102.
- Weng, J.; Li, W.; Jia, X.; An, W. Alleviation of Ischemia-Reperfusion Injury in Liver Steatosis by Augmenter of Liver Regeneration Is Attributed to Antioxidation and Preservation of Mitochondria. Transplantation 2017, 101, 2340–2348.
- Mansouri, A.; Gattolliat, C.H.; Asselah, T. Mitochondrial Dysfunction and Signaling in Chronic Liver Diseases. Gastroenterology 2018, 155, 629–647.
- Lei, Z.; Deng, M.; Yi, Z.; Sun, Q.; Shapiro, R.A.; Xu, H.; Li, T.; Loughran, P.A.; Griepentrog, J.E.; Huang, H.; et al. cGAS-mediated autophagy protects the liver from ischemia-reperfusion injury independently of STING. Am. J. Physiol. Gastrointest. Liver Physiol. 2018, 314, G655–G667.
- Ravingerova, T.; Kindernay, L.; Bartekova, M.; Ferko, M.; Adameova, A.; Zohdi, V.; Bernatova, I.; Ferenczyova, K.; Lazou, A. The Molecular Mechanisms of Iron Metabolism and Its Role in Cardiac Dysfunction and Cardioprotection. Int. J. Mol. Sci. 2020, 21, 7889.
- Panisello-Roselló, A.; Alva, N.; Flores, M.; Lopez, A.; Castro Benítez, C.; Folch-Puy, E.; Rolo, A.; Palmeira, C.; Adam, R.; Carbonell, T.; et al. Aldehyde Dehydrogenase 2 (ALDH2) in Rat Fatty Liver Cold Ischemia Injury. Int. J. Mol. Sci. 2018, 19, 2479.
- Panisello-Rosello, A.; Lopez, A.; Folch-Puy, E.; Carbonell, T.; Rolo, A.; Palmeira, C.; Adam, R.; Net, M.; Rosello-Catafau, J. Role of aldehyde dehydrogenase 2 in ischemia reperfusion injury: An update. World J. Gastroenterol. 2018, 24, 2984–2994.
- Zhang, S.; Feng, Z.; Gao, W.; Duan, Y.; Fan, G.; Geng, X.; Wu, B.; Li, K.; Liu, K.; Peng, C. Aucubin Attenuates Liver Ischemia-Reperfusion Injury by Inhibiting the HMGB1/TLR-4/NF-kappaB Signaling Pathway, Oxidative Stress, and Apoptosis. Front. Pharm. 2020, 11, 544124.
- Ryter, S.W. Therapeutic Potential of Heme Oxygenase-1 and Carbon Monoxide in Acute Organ Injury, Critical Illness, and Inflammatory Disorders. Antioxidants 2020, 9, 1153.
- Jung, S.S.; Moon, J.S.; Xu, J.F.; Ifedigbo, E.; Ryter, S.W.; Choi, A.M.; Nakahira, K. Carbon monoxide negatively regulates NLRP3 inflammasome activation in macrophages. Am. J. Physiol. Lung Cell. Mol. Physiol. 2015, 308, L1058–L1067.
- Stucki, D.; Steinhausen, J.; Westhoff, P.; Krahl, H.; Brilhaus, D.; Massenberg, A.; Weber, A.P.M.; Reichert, A.S.; Brenneisen, P.; Stahl, W. Endogenous Carbon Monoxide Signaling Modulates Mitochondrial Function and Intracellular Glucose Utilization: Impact of the Heme Oxygenase Substrate Hemin. Antioxidants 2020, 9, 652.
- Cannistra, M.; Ruggiero, M.; Zullo, A.; Gallelli, G.; Serafini, S.; Maria, M.; Naso, A.; Grande, R.; Serra, R.; Nardo, B. Hepatic ischemia reperfusion injury: A systematic review of literature and the role of current drugs and biomarkers. Int. J. Surg. 2016, 33 (Suppl. 1), S57–S70.
- Birk, A.V.; Liu, S.; Soong, Y.; Mills, W.; Singh, P.; Warren, J.D.; Seshan, S.V.; Pardee, J.D.; Szeto, H.H. The mitochondrial-targeted compound SS-31 re-energizes ischemic mitochondria by interacting with cardiolipin. J. Am. Soc. Nephrol. 2013, 24, 1250–1261.
- Lu, H.I.; Huang, T.H.; Sung, P.H.; Chen, Y.L.; Chua, S.; Chai, H.Y.; Chung, S.Y.; Liu, C.F.; Sun, C.K.; Chang, H.W.; et al. Administration of antioxidant peptide SS-31 attenuates transverse aortic constriction-induced pulmonary arterial hypertension in mice. Acta Pharm. Sin. 2016, 37, 589–603.
- Gong, W.-H. Coexistence of hyperlipidemia and acute cerebral ischemia/reperfusion induces severe liver damage in a rat model. World J. Gastroenterol. 2012, 18, 4934.
- Boyman, L.; Greiser, M.; Lederer, W.L. Calcium influx through the mitochondrial calcium uniporter holocomplex, MCUcx. J. Mol. Cell. Cardiol. 2021, 151, 145–154.
- Belosludtsev, K.N.; Dubinin, M.V.; Belosludtseva, N.V.; Mironova, G.D. Mitochondrial Ca2+ Transport: Mechanisms, Molecular Structures, and Role in Cells. Biochemistry 2019, 84, 593–607.
- Perocchi, F.; Gohil, V.M.; Girgis, H.S.; Bao, X.R.; McCombs, J.E.; Palmer, A.E.; Mootha, V.K. MICU1 encodes a mitochondrial EF hand protein required for Ca(2+) uptake. Nature 2010, 467, 291–296.
- Li, J.; Li, R.J.; Lv, G.Y.; Liu, H.Q. The mechanisms and strategies to protect from hepatic ischemia-reperfusion injury. Eur. Rev. Med. Pharmacol. Sci. 2015, 19, 2036–2047.
- Chang, W.J.; Chehab, M.; Kink, S.; Toledo-Pereyra, L.H. Intracellular Calcium Signaling Pathways during Liver Ischemia and Reperfusion. J. Investig. Surg. 2010, 23, 228–238.
- Saeed, W.K.; Jun, D.W.; Jang, K.; Chae, Y.J.; Lee, J.S.; Kang, H.T. Does necroptosis have a crucial role in hepatic ischemia-reperfusion injury? PLoS ONE 2017, 12, e0184752.
- Li, W.; Zhang, X.B.; Wu, R.C.; Zhang, S.N.; Liu, J.; Gao, Y.; Zheng, K.P.; Ran, J.H. Calcium-calmodulin-dependent protein kinase type 2 induces apoptosis of hepatocytes after liver transplantation. Eur. Rev. Med. Pharm. Sci. 2020, 24, 3331–3343.
- Nakazato, P.C.G.; Victorino, J.P.; Fina, C.F.; Mendes, K.D.S.; Gomes, M.C.J.; Evora, P.R.B.; D’Albuquerque, L.A.C.; Castro, E.S.O. Liver ischemia and reperfusion injury. Pathophysiology and new horizons in preconditioning and therapy. Acta Cir. Bras. 2018, 33, 723–735.
- Dolder, M.; Walzel, B.; Speer, O.; Schlattner, U.; Wallimann, T. Inhibition of the mitochondrial permeability transition by creatine kinase substrates. Requirement for microcompartmentation. J. Biol. Chem. 2003, 278, 17760–17766.
- Caretti, A.; Bianciardi, P.; Sala, G.; Terruzzi, C.; Lucchina, F.; Samaja, M. Supplementation of creatine and ribose prevents apoptosis in ischemic cardiomyocytes. Cell. Physiol. Biochem. 2010, 26, 831–838.
- Weemhoff, J.L.; Woolbright, B.L.; Jenkins, R.E.; McGill, M.R.; Sharpe, M.R.; Olson, J.C.; Antoine, D.J.; Curry, S.C.; Jaeschke, H. Plasma biomarkers to study mechanisms of liver injury in patients with hypoxic hepatitis. Liver Int. 2017, 37, 377–384.
- Zhao, G.; Shen, X.; Nan, H.; Yan, L.; Zhao, H.; Yu, J.; Lv, Y. Remifentanil protects liver against ischemia/reperfusion injury through activation of anti-apoptotic pathways. J. Surg. Res. 2013, 183, 827–834.
- Kang, J.H.; Lee, H.S.; Park, D.; Kang, Y.W.; Kim, S.M.; Gong, J.R.; Cho, K.H. Context-independent essential regulatory interactions for apoptosis and hypertrophy in the cardiac signaling network. Sci. Rep. 2017, 7, 34.
- Joiner, M.L.; Koval, O.M.; Li, J.; He, B.J.; Allamargot, C.; Gao, Z.; Luczak, E.D.; Hall, D.D.; Fink, B.D.; Chen, B.; et al. CaMKII determines mitochondrial stress responses in heart. Nature 2012, 491, 269–273.
- Zhao, H.; Meng, W.; Li, Y.; Liu, W.; Fu, B.; Yang, Y.; Zhang, Q.; Chen, G. The protective effects of CHIR99021 against oxidative injury in LO2 cells. Pharmazie 2016, 71, 629–635.
- Gracia-Sancho, J.; Casillas-Ramírez, A.; Peralta, C. Molecular pathways in protecting the liver from ischaemia/reperfusion injury: A 2015 update. Clin. Sci. 2015, 129, 345–362.
- Calvert, J.W.; Jha, S.; Gundewar, S.; Elrod, J.W.; Ramachandran, A.; Pattillo, C.B.; Kevil, C.G.; Lefer, D.J. Hydrogen sulfide mediates cardioprotection through Nrf2 signaling. Circ. Res. 2009, 105, 365–374.
- Fu, H.; Chen, H.; Wang, C.; Xu, H.; Liu, F.; Guo, M.; Wang, Q.; Shi, X. Flurbiprofen, a cyclooxygenase inhibitor, protects mice from hepatic ischemia/reperfusion injury by inhibiting GSK-3beta signaling and mitochondrial permeability transition. Mol. Med. 2012, 18, 1128–1135.
More