1. Introduction
Mycotoxins are toxic secondary metabolites produced by many filamentous fungi of ascomycetes
[1]. Mycotoxin pollution is a persistent global problem which is inevitable and unpredictable. The production of mycotoxins is affected by the surrounding environment; even a good growth and storage environment cannot completely prevent the production of mycotoxins
[2]. Fumonisins are a group of toxins that pose a significant threat to food and animal health after aflatoxins. Fumonisins have high toxicity and often appear together with aflatoxin toxicity. They cause huge economic losses to the livestock and poultry breeding industry and threaten human health
[3][4][3,4]. Therefore, several studies have been exploring methods to control and alleviate fumonisin toxicity. Fumonisins easily contaminate corn, rice, and other grains, causing damage to the liver and kidneys of several animals that feed on these grains and even causing tumor problems
[5][6][5,6]. In addition, fumonisin toxicity is implicated in causing human esophageal cancer and neural tube defect disease
[7][8][7,8], thus fumonisins have gradually become a research hotspot after aflatoxin.
Fumonisins are a water-soluble secondary metabolite mainly produced by
Fusarium verticillioides,
Fusarium proliferatum, and other
Fusarium species
[9]. It exists on a variety of substrates, mainly on grains such as corn, and can also be found in products manufactured using grains as raw materials
[5]. Fumonisins can be divided into four categories: A, B, C and P, including 28 structural analogues: FA
1, FA
2, FA
3, PHFA
3a, PHFA
3b, HFA
3, FAK
1, FBK
1, FB
1, Iso-FB
1, PHFB
1a, PHFB
1b, HFB
1, FB
2, FB
3, FB
4, FB
5, FC
1, N-acetyl-FC
1, Iso-FC
1, N-acetyl-iso-FC
1, OH-FC
1, N-acetyl-OH-FC
1, FC
3, FC
4, FP
1, FP
2, and FP
3. Notably, the fumonisin B family is the main and most toxic family. Fumonisin B
1 (FB
1) and fumonisin B
2 (FB
2) are the most abundant and most toxic variants that naturally contaminate maize, accounting for 70–80% and 15–25% of the total number of fumonisins
[10][11][10,11].
WHO (2001) established a provisional maximum daily tolerable level of fumonisins at 2 μg/kg-BW (body weight), owing to its high levels and high toxicity
[12]. The European Commission (2006 and 2007) set the maximum levels of fumonisins for unprocessed maize at 4000 μg/kg, FB at 1000 μg/kg for human corn-based foods, 800 μg/kg for corn breakfast cereals and snacks, and 200 μg/kg for corn-based baby foods
[13][14][13,14]. The International Agency for Research on Cancer (IARC) classifies fumonisins into group 2B, which is a possible human carcinogen owing to their harmful effects
[15]. Therefore, it is particularly significant to reduce the content and detoxify fumonisins in food.
Fumonisins are highly soluble in water and have strong thermal stability, thus they are chemically stable under various conditions. It is therefore challenging to remove them from ordinary grain processing to meet normal edible standards
[16]. Physical and chemical methods cannot effectively remove fumonisins and other toxic substances from grains. Studies report that biological methods can effectively remove fumonisins in crops. Therefore, studies have widely explored the inhibition of fumonisin-producing strain growth and degradation of fumonisins through biological control and biodegradation
[17][18][17,18].
2. Effect of Fumonisins on Sphingolipid Synthesis
Fumonisins (such as FB
1) are a class of structurally similar diesters comprising different polyols and glycerol tricarboxylic acids. They have a similar structure to that of sphingosine (So) and sphinganine (Sa). Therefore, they are classified under sphingosine-like mycotoxins
[19][20][21,22]. So and Sa are the main components of sphingomyelin. Sphingolipid is an important component of biofilm. Sphingolipid is involved in the regulation of several signal transduction processes such as cell proliferation, differentiation, senescence, apoptosis, and carcinogenesis. Notably, sphingolipid is the key hub of cell-to-cell recognition and interaction
[21][22][23][23,24,25].
So and Sa are biosynthesized as condensation between palmitoyl-COA and serine as substrates under the actions of serine palmitoyltransferase, ketoreductase, dihydroceramide synthetase, dihydroceramide dehydrogenase, and other enzymes under normal physiological conditions. FB
1 competes and inhibits ceramide synthetase owing to the similar structure to that of So and Sa. Ceramide can be produced through ab initio synthesis, or through sphingomyelin hydrolysis and sphingomyelin circulation, which is the central link of sphingomyelin metabolism. Dysregulation of ceramide synthesis affects sphingomyelin metabolism
[20][21][22,23], resulting in damage to the integrity of the cell membrane. Accumulation of phosphorylated products of So, Sa and So, Sa, sphingosine-1-phosphate (So-1-p) and sphinganin-1-phosphate (Sa-1-p) in cells can also cause cellular dysfunction
[24][19].
The contents of Sa and So in blood and cerebrospinal fluid of pigs were significantly higher compared with those in the control group after intravenous administration of 139 nmol of FB
1 or oral administration of 3425 nmol/kg-BW FB
1 [19][21], therefore, Sa and So are biomarkers of FB
1 exposure in vivo. Waes et al. reported that the content of Sa-1-p in plasma of LM/Bc mice treated with 40 μM FB
1 was significantly higher relative to that of the control group
[25][26], This dose of FB
1 increases the rate of embryonic malformation and the risk of neural tube defects in pregnant LM/BC mice
[26][27]. Moreover, Kim et al. reported that accumulation of Sa-1-p and So-1-p in serum was significant compared with that of Sa and So after intraperitoneal administration of 10 mg/kg FB
1 in mice for five consecutive days
[27][28]. In addition, the kidney is the main metabolic organ of FB
1; Sa-1-p, and So-1-p produced by FB
1 metabolism accumulate and last longer in the kidney relative to FB
1, and So-1-p and Sa-1-p in cells can cause cellular dysfunction, implying that Sa-1-p and So-1-p can also be used as biomarkers for FB
1 exposure in the body. This explains why the kidney is more vulnerable to injury compared with other organs
[28][29][29,30]. Grenier et al. reported that fumonisins cause changes in the contents of Sa and So and exert a significant increase in levels of pro-inflammatory factors and Th1/Th7 in the small intestine with the increase in fumonisin concentration and feeding time
[30][31]. Bracarense et al. reported similar findings when studying the effect of FB
1 on the expression of pro-inflammatory cytokines mRNA in porcine jejunal epithelial cells. They found that the expression of IFN- γ and IL-10 in porcine jejunal epithelial cells increased significantly
[31][32]. These findings indicate that FB
1 affects sphingolipid metabolism and exerts cytotoxicity by modulating the expression of proinflammatory cytokines.
3. Fumonism Induces Oxidative Stress
Oxidants and antioxidants in the body are in a state of dynamic balance under physiological conditions. The body produces potentially excessive toxic aerobic free radicals, on exposure to FB
1, which cannot be neutralized by antioxidants present in cells. High levels of aerobic free radicals result in lipid peroxidation, DNA oxidative damage, decreased glutathione (GSH) content, and the down-regulated expression of glutathione peroxidase (GPx) and superoxide dismutase (SOD), ultimately leading to cell tissue damage and dysfunction
[32][33][34][35][36][37][33,34,35,36,37,38].
Studies report that FB
1 mediates cytotoxicity partially through the induction of oxidative stress. For example, treatment of HepG2 cells with 50 μM FB
1 for 0, 12, and 24 h, significantly increased the ROS content in HepG2 cells treated with FB
1 compared with the level of the control group
[37][38]. The levels of SOD-1, SOD-2, glutathione reductase (GR), and catalase (CAT) in the colon tissue of mice exposed to 2.5 mg/kg-BW FB
1 for 4 consecutive days were significantly lower relative to the levels of the control group. On the contrary, expression levels of CYP450, thioredoxin, heat shock protein 70, and heat shock protein 90 were significantly higher relative to the expression levels of the control group
[38][20]. Exposure of human SH-SY5Y neuroblastoma, rat C6 glioblastoma, and mouse GT1-7 hypothalamic cells to 0.1–100 μM FB
1 for 0–144 h induced C6 and GT1-7 ROS formation in a dose-dependent manner, however, it had no significant effect on SH-SY5Y. Moreover, it downregulated GSH expression, increased malondialdehyde (MDA) production, and promoted lipid peroxidation and necrotic cell death in all cells
[35][36].
In addition to oxidative stress, FB
1 can cause damage to cell DNA. Domijan et al. observed that the apoptosis rate was significantly increased in adult male rats 48 h after administration of 5 μg/kg-BW FB
1, in a dose-dependent manner. Furthermore, treatment of adult male rats with 500 μg/kg-BW FB
1 induced significant DNA damage
[39]. DNA damage is the basis of cell carcinogenesis. Treatment of frozen horse spermatozoa with 2.5 × 10
−5 μM FB
1 showed significant damage to sperm chromosomes resulting in reproductive toxicity
[40]. Exposure of C6 glioma cells and p53 deleted mouse embryonic fibroblasts to FB
1 significantly increased the content of MDA in C6 glioma cells, increased the apoptotic rate of C6 glioma cells, and increased the levels of 8-OH-dG and DNA fragments in C6 glioma cells in a dose-dependent manner
[41]. 8-OH-dG is an important marker for DNA oxidative damage and cell carcinogenesis
[42][43][42,43]. This finding indicates that FB
1 leads to DNA oxidative damage. In addition, Yuan et al. reported that exposure of pig iliac endothelial cells to 50 µg/mL FB
1 induced a significant increase in intracellular MDA content and a decrease in SOD, CAT, and GSH levels. Moreover, the findings showed that FB
1 affects the expression of porcine vascular endothelial cells’ tight junction proteins
[44]. Yu et al. conducted a subsequent study and reported that FB
1 promotes cell proliferation and migration as well as induces carcinogenesis of human esophageal epithelial cells. Notably, FB
1 significantly upregulates the expression of cell cycle regulatory proteins (cyclinD1 and cyclinD3) and downregulates the expression of tumor suppressor genes such as phosphatase, tensin homolog, and adenomatous polyposis, indicating that FB
1 may exert its toxic or carcinogenic effects by modulating the cell cycle
[45].
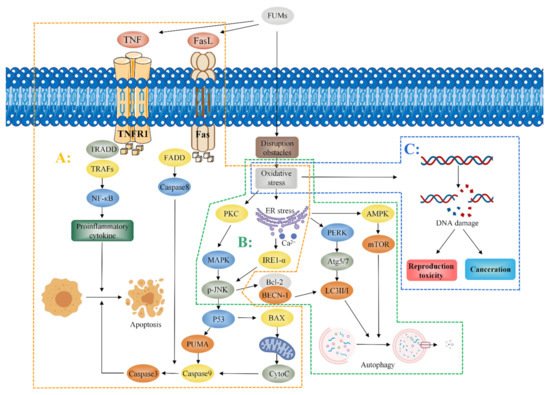
Oxidative stress induced by FB
1 partially mediates apoptosis and autophagy. Mitogen-activated protein kinase (MAPK) is an important messenger in cells. When FB
1 is administered to cells, it activates protein kinase C (PKC) and regulates the c-Jun N-terminal kinase (JNK) signal pathway through MAPK
[46][47][46,47]. Administration of 2.5 mg/kg-BW FB
1 to mice for 4 days induced oxidative stress and the endoplasmic reticulum release of Ca
2+, resulting in JNK phosphorylation, activation of the p53 apoptosis signal pathway, upregulation of the expression of pro-apoptotic factors (PUMA and Caspase3), and the induction of apoptosis
[38][20]. Studies report that mitochondria also play a role in apoptosis
[48]. Khan et al. observed that mitochondria induce the production of cytochrome C (CytoC) by p53-activated BAX. BAX is a member of the Bcl-2 family of pro-apoptotic proteins which promotes the expression of Caspase9, as well as the expression of Caspase3, ultimately inducing cell apoptosis
[49]. Notably, FB
1 may induce apoptosis through the Fas/FasL pathway. FB
1 promotes dysfunction of the Fas receptor and activation of the caspase8 pathway inducing Caspase3 expression and resulting in cell apoptosis
[50][51][50,51]. Kim et al. reported that oxidative stress mediates the JNK pathway through the effects on the endoplasmic reticulum, as well as promotes interaction between Bcl-2 and BECN-1 to release BECN1. BECN1 is a key regulator of autophagy. BECN1 modulates expression of the autophagy-associated protein LC3-II/I to induce autophagy and promotes autophagy through the endoplasmic reticulum-mediated expression of Inositol-requiring enzyme-1-α (IRE1-α), PERK-induced ATG5, ATG7, and LC3-II/I
[38][20]. Notably, exposure to FB
1 upregulates the expression of AMP-dependent protein kinase (AMPK) and downregulates expression of mammalian target of rapamycin (mTOR) by mediating endoplasmic reticulum stress, thus inducing autophagy
[37][38]. Moreover, tumor necrosis factor-alpha (TNF-α) plays an important role in the toxicity induced by FB
1. He et al. reported that the activity of TNF-α and expression level of TNF-α mRNA in heart and lung tissues of mice increased after a subcutaneous administration of 2.25 mg/kg-BW FB
1 to male and female BALB/c mice for 5 days
[52]. KÓCSÓ et al. reported similar findings that FB
1 upregulates TNF-α mRNA expression and increases the activity of TNF-α
[53]. Chen et al. reported that FB
1 upregulated the expression of TNF-α mRNA in PK-15 cells and induced apoptosis in porcine kidney cells PK-15, indicating that TNF-α can be used as a biomarker for FB
1 exposure in vivo
[54]. Régnier et al. observed that administration of 10 mg/kg FB
1 through diet upregulated the expression of NF-κB and Interleukin-8 in the liver and jejunum. Notably, NF-κB is an important target in the TNF signal pathway, implying that TNF-α may play a role in the toxicity induced by FB
1 [55]. The mechanism of fumonisin toxicity is presented in
Figure 1.
Figure 1. Mechanism of fumonisin toxicity. Fumonisins induce oxidative stress and mediate apoptosis, autophagy, reproductive toxicity, and promote carcinogenesis. A: The apoptosis pathway. FB1 activates PKC, which activates the JNK signaling pathway through MAPK. Meanwhile, FB1 induces oxidative stress, resulting in the release of Ca2+ from the endoplasmic reticulum; increased Ca2+ release leads to phosphorylation of JNK, which further activates the P53 apoptosis signaling pathway. P53 up-regulates the expressions of pro-apoptotic factors PUMA and Caspase3 and activates BAX, which induces increased CytoC expression. It promotes the expression of Caspase9 and Caspase3 and induces apoptosis. FB1 also leads to Fas receptor dysfunction, which activates Caspase8, and Caspase8 induces increased expression of Caspase3, leading to apoptosis; FB1 induces apoptosis by activating the TNF-α signaling pathway and upregulating NF-κB, an important target of the TNF-α signaling pathway. B: The autophagy pathway. FB1 mediates the JNK pathway through oxidative stress, promotes the interaction between BAL-2 and BECN-1, and induces increased LC3-II/I expression. Meanwhile, FB1 mediates PERK through the endoplasmic reticulum, induces ATG5/7 expression, increases LC3-II/I expression, and induces apoptosis. FB1 also up-regulates the AMP-dependent AMPK expression through endoplasmic reticulum stress and increased AMPK expression down-regulates the mTOR induced autophagy. C: Reproductive toxicity and carcinogenesis. Oxidative stress induces DNA damage which in turn induces reproductive toxicity and carcinogenesis.