Despite the great strides in healthcare during the last century, some challenges still remained unanswered. The development of multi-drug resistant bacteria, the alarming growth of fungal infections, the emerging/re-emerging of viral diseases are yet a worldwide threat. Since the discovery of natural antimicrobial peptides able to broadly hit several pathogens, peptide-based therapeutics have been under the lenses of the researchers. This review aims to focus on synthetic peptides and elucidate their multifaceted mechanisms of action as antiviral, antibacterial and antifungal agents. Antimicrobial peptides generally affect highly preserved structures, e.g., the phospholipid membrane via pore formation or other constitutive targets like peptidoglycans in Gram-negative and Gram-positive bacteria, and glucan in the fungal cell wall. Additionally, some peptides are particularly active on biofilm destabilizing the microbial communities. They can also act intracellularly, e.g., on protein biosynthesis or DNA replication. Their intracellular properties are extended upon viral infection since peptides can influence several steps along the virus life cycle starting from viral receptor-cell interaction to the budding. Besides their mode of action, improvements in manufacturing to increase their half-life and performances are also taken into consideration together with advantages and impairments in the clinical usage. Thus far, the progress of new synthetic peptide-based approaches is making them a promising tool to counteract emerging infections.
- antimicrobial peptides
- antifungal
- antibacterial
- antiviral
- peptide-based therapies
- synthetic peptides
1. Introduction
All antimicrobial peptides (AMPs) share common features, such as a sequence composed of less than 100 amino acids (aa), [1] with the majority having between 10 and 60 aa [2]. Even if some anionic AMPs, rich in glutamic and aspartic acids, are negatively charged [3], almost all antimicrobial peptides have a net positive charge for the presence of a high number of lysine, arginine and histidine (protonated in acidic conditions) [4]. Finally, another common feature is represented by the hydrophobicity conferred by hydrophobic aa that often overcomes 50% of the total amino acid sequence [5]. The high lipophilicity is useful especially for the penetration in the biological membranes but considering the net charge, overall, AMPs are amphipathic molecules. The classifications are based on their structure or the presence/absence of recognizable motifs. AMPs could be α-helix, β-sheet, linearly extended, both α-helix and β-sheet, cyclic and with complex structure or, seen from a different perspective, tryptophan- and arginine-rich, histidine-rich, proline-rich and glycine-rich [6][7].
In the last decades, the increasing resistance to antibiotic treatments, i.e., Methicillin, Vancomycin-resistant Staphilococus aureus and the rise of species with intrinsic multi-drug resistance, such as Candida auris, highlights the need for the development of new agents [8][9][10]. Studies on the AMPs synthetic analogs provided a new tool to understand the different and unique modes of actions against diverse microorganisms.
2. Synthetic Antimicrobial Peptides
3. Antibacterial Peptides and Their Mechanism of Action
Many factors can influence membrane perturbation and disruption by AMPs, i.e., amino acids sequence, the lipid composition of the membrane, peptide concentration as well as differences in membrane composition between eukaryotic and bacterial cells allow the AMPs to distinguish a microbial target from the host. Bacterial membranes are negatively charged due to the presence of anionic phospholipids groups, e.g., phosphatidylglycerol, phosphatidylserine, while eukaryotic cells possess groups with a neutral charge, e.g., phosphatidylcholine and phosphatidylethanolamine [39]. Moreover, the presence of cholesterol, a common feature in eukaryotic cells, is able to interact with AMPs either neutralizing or reducing their activity or stabilizing the phospholipid bilayer [40].
In Gram-positive bacteria, AMPs have to cross first the cell wall composed of crosslinked peptidoglycan with lipoteichoic acid prior to reaching the membrane whereas in Gram-negative they face a coat of lipopolysaccharide (LPS) followed by a phospholipidic outer membrane and a less cross-linked peptidoglycan layer [41]. Electrostatic interactions between the cationic peptide and the negatively charged components, e.g., lipopolysaccharide in Gram-negative and teichoic acid in Gram-positive, are the first steps to contribute to bacterial membrane affinity [42]. However, while AMPs seem to traverse the peptidoglycan layer with ease and access to the cytoplasmic membrane of the Gram-positive, they need to disrupt or perturb both outer and cytoplasmic membrane in Gram-negatives. Impedance in crossing or permeabilization results in loss of antimicrobial activity (Figure 1a (A,B)) [43].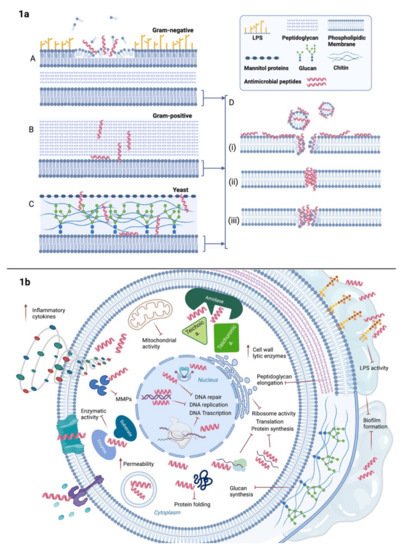 Figure 1. AMPs broad-spectrum antimicrobial activity. (a) Primarily, AMPs’action is based on their action on cytoplasmic membranes, i.e., perturbation or disruption. However, in presence of Gram-negative bacteria (A) AMPs have to firstly cross the outer phospholipidic membrane and secondly traverse the peptidoglycan layer before reaching the inner membrane. In Gram-positive bacteria (B) they navigate through the thick cell wall of peptidoglycan and in fungi (C), they encounter mannitol proteins, glucans and chitin prior to access to the cytoplasmic membrane. Once reached the phospholipidic bilayer, they induce perturbation via pore formation following either (D) (i) carpet-like, (ii) barrel-stave or (iii) or toroidal pore model depending on the peptide composition. (b) Besides pore formation, some AMPs bind some components and receptors on the extracellular side of the membrane, i.e., Toll-like receptors; others manage to enter the cytosol through direct penetration in vesicles or channels thus destabilizing the permeability and activating the inflammatory cytokines cascade. Intracellularly, they could also interfere with DNA or RNA leading to degradation and cell death. They may also affect mitochondrial activity or protein synthesis by targeting ribosome subunits or protein folding. In the case of bacterial cell wall, they can prevent elongation of peptidoglycan chains or hinder teichoic and teichuronic binding acids to amidases. Cell wall components inhibition will promote cell autolysis. In the extracellular space, AMPs can sequestrate LPS reducing the impact of endotoxins on the host’s immune response. In fungal cells, AMPs can intervene on glucan synthesis thus blocking the building pieces of their wall. Further inhibitory action on biofilm matrix impairs the quorum sensing and improves the susceptibility of the single pathogens in both bacterial and fungal communities.
In order to explain the perturbation of the phospholipidic membranes operated by the AMPs, three main models have been proposed: carpet-like, barrel-stave and toroidal pore (Figure 1a (D)). Generally, when the ratio of peptide/lipids is low, AMPs interact with the phospholipidic layer of the membrane in a parallel manner, defined as carpet-like model, and interaction among the peptides or penetration in the hydrophobic core of the bilayer are not taking place [44]. Membrane integrity is disrupted and micelles are formed as in a detergent-like process [45]. With increasing AMPs ratio, they move to a perpendicular orientation until reaching such a concentration that they can cross the membrane forming pores (1:50–1:500 and more) [46][47]. A minimum length of ~22 amino acid for α-helix peptides is required to span the phospholipid layer, while β-sheet structures necessitate a minimum of 8 [48].
In the barrel-stave, interaction among peptides is a prerequisite as they mimic a transmembrane pore, whereas, in the case of the toroidal model, peptides are loosely arranged [49][50]. Despite the perturbation of the membrane seems to vary depending on the peptides, actually, the mechanisms of action are not completely well-defined and they are partially overlapping [51]. Moreover, all these models are based on the membrane perturbation but, then, the killing effect is not always enough to provide antimicrobial activity [52].
Besides membrane disruption, recent studies showed how peptides could act on other targets as well (Figure 1b) [53]. Some AMPs have shown their efficacy by binding some components and receptors on the extracellular side of the membrane and wall, thus destabilizing the permeability and/or activating intracellular signaling pathways that have, as a response, the inhibition or the activation of several functions.
The inhibitors of the nucleic acid biosynthesis seem to have a high binding affinity for both DNA and RNA because they share with nucleic acid-binding enzymes or substrates, homologous fragments of their sequences; an interesting example is represented by DNA-binding protein histone H2A [54]. Other mechanisms use the inhibition of the enzymes involved in the DNA/RNA biosynthesis, like DNA topoisomerase I preventing DNA relaxation [55], RNA polymerase blocking the transcription [56] and gyrase impairing the supercoiling of DNA. [57] As a result, DNA/RNA degradation is induced and consequentially also cell death. There are several inhibitors of protein biosynthesis which alter the transcription and the translation but also the correct folding and the degradation of the protein. Usually, the AMPs that act on the protein biosynthesis target the ribosome subunits [58] but some others can interfere with the incorporation of histidine, uridine and thymidine [59][60], the amino acid synthesis pathways [61], the release factors on the ribosome [62], the regulation of sigma factors [63], the nucleotide and coenzyme transport [61] and the degradation of DNA-replication-associated proteins [64]. Some peptides influence protein folding, in particular, DnaK, the major Hsp70 of the chaperone pathway in Escherichia coli, which has been seen as an optimal target to prevent the refolding of misfolded proteins [65]. Another approach is linked to the inhibition of matrix metalloproteases, essential enzymes in microbial cell growth and homeostasis, i.e., serine protease, trypsin-like protease, elastase and chymotrypsins [66][67][68]. There are also inhibitors of cell division that block DNA replication or the mechanisms essential for the repair of DNA damages, then resulting in the block of the cell cycle, in the impairment of the chromosome separation, in the failure of septation, in the alteration of mitochondrial activity and in a substantial change in the cell morphology with clearly visible blebbing and elongation towards a filamentous shape [69][70].
Cell wall synthesis is another suitable target. Some AMPs act on lipid II by sequestrating it from the functional site [71][72] or by binding D-Ala-D-Ala residues of its precursor preventing the addition of N-acetylglucosamine and N-acetylmuramic acid in the structure, hence the peptidoglycan elongation [73].
Figure 1. AMPs broad-spectrum antimicrobial activity. (a) Primarily, AMPs’action is based on their action on cytoplasmic membranes, i.e., perturbation or disruption. However, in presence of Gram-negative bacteria (A) AMPs have to firstly cross the outer phospholipidic membrane and secondly traverse the peptidoglycan layer before reaching the inner membrane. In Gram-positive bacteria (B) they navigate through the thick cell wall of peptidoglycan and in fungi (C), they encounter mannitol proteins, glucans and chitin prior to access to the cytoplasmic membrane. Once reached the phospholipidic bilayer, they induce perturbation via pore formation following either (D) (i) carpet-like, (ii) barrel-stave or (iii) or toroidal pore model depending on the peptide composition. (b) Besides pore formation, some AMPs bind some components and receptors on the extracellular side of the membrane, i.e., Toll-like receptors; others manage to enter the cytosol through direct penetration in vesicles or channels thus destabilizing the permeability and activating the inflammatory cytokines cascade. Intracellularly, they could also interfere with DNA or RNA leading to degradation and cell death. They may also affect mitochondrial activity or protein synthesis by targeting ribosome subunits or protein folding. In the case of bacterial cell wall, they can prevent elongation of peptidoglycan chains or hinder teichoic and teichuronic binding acids to amidases. Cell wall components inhibition will promote cell autolysis. In the extracellular space, AMPs can sequestrate LPS reducing the impact of endotoxins on the host’s immune response. In fungal cells, AMPs can intervene on glucan synthesis thus blocking the building pieces of their wall. Further inhibitory action on biofilm matrix impairs the quorum sensing and improves the susceptibility of the single pathogens in both bacterial and fungal communities.
In order to explain the perturbation of the phospholipidic membranes operated by the AMPs, three main models have been proposed: carpet-like, barrel-stave and toroidal pore (Figure 1a (D)). Generally, when the ratio of peptide/lipids is low, AMPs interact with the phospholipidic layer of the membrane in a parallel manner, defined as carpet-like model, and interaction among the peptides or penetration in the hydrophobic core of the bilayer are not taking place [44]. Membrane integrity is disrupted and micelles are formed as in a detergent-like process [45]. With increasing AMPs ratio, they move to a perpendicular orientation until reaching such a concentration that they can cross the membrane forming pores (1:50–1:500 and more) [46][47]. A minimum length of ~22 amino acid for α-helix peptides is required to span the phospholipid layer, while β-sheet structures necessitate a minimum of 8 [48].
In the barrel-stave, interaction among peptides is a prerequisite as they mimic a transmembrane pore, whereas, in the case of the toroidal model, peptides are loosely arranged [49][50]. Despite the perturbation of the membrane seems to vary depending on the peptides, actually, the mechanisms of action are not completely well-defined and they are partially overlapping [51]. Moreover, all these models are based on the membrane perturbation but, then, the killing effect is not always enough to provide antimicrobial activity [52].
Besides membrane disruption, recent studies showed how peptides could act on other targets as well (Figure 1b) [53]. Some AMPs have shown their efficacy by binding some components and receptors on the extracellular side of the membrane and wall, thus destabilizing the permeability and/or activating intracellular signaling pathways that have, as a response, the inhibition or the activation of several functions.
The inhibitors of the nucleic acid biosynthesis seem to have a high binding affinity for both DNA and RNA because they share with nucleic acid-binding enzymes or substrates, homologous fragments of their sequences; an interesting example is represented by DNA-binding protein histone H2A [54]. Other mechanisms use the inhibition of the enzymes involved in the DNA/RNA biosynthesis, like DNA topoisomerase I preventing DNA relaxation [55], RNA polymerase blocking the transcription [56] and gyrase impairing the supercoiling of DNA. [57] As a result, DNA/RNA degradation is induced and consequentially also cell death. There are several inhibitors of protein biosynthesis which alter the transcription and the translation but also the correct folding and the degradation of the protein. Usually, the AMPs that act on the protein biosynthesis target the ribosome subunits [58] but some others can interfere with the incorporation of histidine, uridine and thymidine [59][60], the amino acid synthesis pathways [61], the release factors on the ribosome [62], the regulation of sigma factors [63], the nucleotide and coenzyme transport [61] and the degradation of DNA-replication-associated proteins [64]. Some peptides influence protein folding, in particular, DnaK, the major Hsp70 of the chaperone pathway in Escherichia coli, which has been seen as an optimal target to prevent the refolding of misfolded proteins [65]. Another approach is linked to the inhibition of matrix metalloproteases, essential enzymes in microbial cell growth and homeostasis, i.e., serine protease, trypsin-like protease, elastase and chymotrypsins [66][67][68]. There are also inhibitors of cell division that block DNA replication or the mechanisms essential for the repair of DNA damages, then resulting in the block of the cell cycle, in the impairment of the chromosome separation, in the failure of septation, in the alteration of mitochondrial activity and in a substantial change in the cell morphology with clearly visible blebbing and elongation towards a filamentous shape [69][70].
Cell wall synthesis is another suitable target. Some AMPs act on lipid II by sequestrating it from the functional site [71][72] or by binding D-Ala-D-Ala residues of its precursor preventing the addition of N-acetylglucosamine and N-acetylmuramic acid in the structure, hence the peptidoglycan elongation [73].
4. AMPs—Goods vs. Bads, and the Long Way towards Clinical Application
There are obvious, multiple advantages of AMPs over classical antibiotics. AMPs are easy to synthesize, thanks to recent advances in automated protein synthesis, or can alternatively be produced in large quantities in heterologous expression systems, either in microbial cells or in plants [74]. In addition, AMPs are largely prone to chemical modification, aimed at overcoming inherent problems, such as susceptibility to enzymatic degradation, chemical/physical instability and toxicity to host cells, thus optimizing molecules’ features and smoothing their pathway towards the clinics [75]. Broad-spectrum activity and rapid killing are other much-appreciated characteristics. Finally, AMPs are increasingly seen as a promising therapeutic alternative for treating biofilm-associated infections, one of the major threats in the field of bacterial infections [76]. A suitable instance of both the limitations to therapeutic use inherent to the nature itself of AMPs and the ways to overcome these is offered by the recent study of Wang Manchuriga and colleagues on temporins [77]. As many natural AMPs isolated from the skin of anuran amphibians (frogs and toads), temporins display a potent antimicrobial activity but this quality is often thwarted by elevated cytotoxicity, in particular against erythrocytes [78]. Working on temporin-GHa from Hylarana guentheri, Manchuriga and colleagues designed several analogs of the naturally-occurring sequence, modifying the type, position and number of charged residues. Some of the derived peptides displayed a significant reduction of hemolytic activity with respect to parent peptide while retaining potent antibacterial activity, but it was not possible to reduce cytotoxicity to zero without compromising antibacterial activity, confirming that a delicate balance of charge and other physico-chemical parameters (e.g., amphipathic and extension of hydrophobic surfaces) is necessary to obtain a plausible therapeutic lead [77]. One of the aspects that are often quoted in support of the (potential) use of AMPs in clinical practice is their low tendency to evoke antibiotic resistance. This tenet stems from the fact that AMPs generally (but not always, as specified above) hit the lipid component of the plasma membrane, a cellular component that is believed per se to be not easily modifiable in its basic physicochemical features by microbial targets. Although the slower emergence of resistance to AMPs with respect to conventional antibiotics is a reality, however, experience and much work have clearly shown that the reassuring thought that the complex phenomenon of resistance would not eventually thwart AMPs’ value, is somewhat naïve and misleading. In fact, the long coevolution of microorganisms and AMPs has spurred the development of several resistance mechanisms. These include sequestration by bacterial enzymes, proteolytic degradation of peptides, efflux pumps to remove AMPs from the periplasmic space, alteration of components of bacterial surface to reduce surface attachment and permeability, down-regulation by immunomodulation [79][80][81][82]. Despite the limitations briefly outlined above, that have hampered their development in the classical drug discovery pipeline, AMPs are attracting continuous and ever-increasing interest as new antimicrobials agents. Out of some ~3000 molecules that have been isolated from different sources, just a handful have been the object of preclinical studies and further proceeded to clinical trials [82]. A recent analysis of AMPs patents from 2015 through 2020 has confirmed a long-standing trend, i.e., the fact that AMPs earmarked for clinical development are in vast majority analogs or derivatives of natural peptides, obtained through a template-based strategy aimed at enhancing the activity and stability of natural AMPs while reducing their toxicity [83]. Currently, just three AMPs have been approved by the U.S. Food and Drug Administration (FDA) for therapeutic use, i.e., gramicidin, colistin and daptomycin. Gramicidin has a long history. First isolated from Bacillus brevis over 70 years ago, gramicidin is active against a range of Gram-positive and Gram-negative bacteria, although its severe toxicity for human erythrocytes has a limited clinical indication to topical applications [84]. Polymyxin and colistin, which are cationic peptides in use for decades, have regained interest lately, due to their strong activity against multi-drug resistant Gram-negative pathogens. Their ability to bind the lipid A component of LPS makes them precious, the last resource weapons to fight septic shock, notwithstanding their known nephrotoxicity. Resistance has emerged, however, and is spreading at an alarming pace, putting the effectiveness of these valuable therapeutics at risk [85][86]. Last but not least, daptomycin. This membrane-active cyclic lipopeptide has received the green light from the FDA in 2003 to treat Gram-positive infections. It is believed that its mechanism of action differs from that of other AMPs since daptomycin causes bacterial membrane depolarization rather than membrane disruption and pore formation [87]. In recent years, resistance in Staphylococcus aureus has been more and more frequently reported, and the search for substitutes that might prolong the clinical use of this important antibiotic is actively underway [88].5. Conclusions
References
- Wang, G. Antimicrobial Peptides: Discovery, Design and Novel Therapeutic Strategies, 2nd ed.; CABI: Wallingford, UK, 2017; ISBN 978-1-78639-039-4.
- Huan, Y.; Kong, Q.; Mou, H.; Yi, H. Antimicrobial Peptides: Classification, Design, Application and Research Progress in Multiple Fields. Front. Microbiol. 2020, 11, 2559.
- Harris, F.; Dennison, S.R.; Phoenix, D.A. Anionic Antimicrobial Peptides from Eukaryotic Organisms. Curr. Protein Pept. Sci. 2009, 10, 585–606.
- López Cascales, J.J.; Zenak, S.; García de la Torre, J.; Lezama, O.G.; Garro, A.; Enriz, R.D. Small Cationic Peptides: Influence of Charge on Their Antimicrobial Activity. ACS Omega 2018, 3, 5390–5398.
- Hancock, R.E.W.; Scott, M.G. The Role of Antimicrobial Peptides in Animal Defenses. Proc. Natl. Acad. Sci. USA 2000, 97, 8856–8861.
- Mishra, A.K.; Choi, J.; Moon, E.; Baek, K.-H. Tryptophan-Rich and Proline-Rich Antimicrobial Peptides. Molecules 2018, 23, 815.
- The Vast Structural Diversity of Antimicrobial Peptides|Elsevier Enhanced Reader. Available online: https://reader.elsevier.com/reader/sd/pii/S0165614719300896?token=AC26A2A3A45C3A5FDA55579937D526F726B0848E72EAAF15FBC5BC9E4C015C21A91A1FDE71DE83D781B6A91FB50B08CE&originRegion=eu-west-1&originCreation=20211028065806 (accessed on 28 October 2021).
- Friedman, D.Z.P.; Schwartz, I.S. Emerging Fungal Infections: New Patients, New Patterns, and New Pathogens. J. Fungi 2019, 5, 67.
- Lockhart, S.R.; Guarner, J. Emerging and Reemerging Fungal Infections. Semin. Diagn. Pathol. 2019, 36, 177–181.
- Lima, P.G.; Oliveira, J.T.A.; Amaral, J.L.; Freitas, C.D.T.; Souza, P.F.N. Synthetic Antimicrobial Peptides: Characteristics, Design, and Potential as Alternative Molecules to Overcome Microbial Resistance. Life Sci. 2021, 278, 119647.
- Hancock, R.E. Cationic Peptides: Effectors in Innate Immunity and Novel Antimicrobials. Lancet Infect. Dis. 2001, 1, 156–164.
- Sarkar, T.; Chetia, M.; Chatterjee, S. Antimicrobial Peptides and Proteins: From Nature’s Reservoir to the Laboratory and Beyond. Front. Chem. 2021, 9, 691532.
- Giuliani, A.; Rinaldi, A. Beyond Natural Antimicrobial Peptides: Multimeric Peptides and Other Peptidomimetic Approaches. Cell. Mol. Life Sci. 2011, 68, 717.
- Nordström, R.; Malmsten, M. Delivery Systems for Antimicrobial Peptides. Adv. Colloid Interface Sci. 2017, 242, 17–34.
- Marr, A.K.; Gooderham, W.J.; Hancock, R.E. Antibacterial Peptides for Therapeutic Use: Obstacles and Realistic Outlook. Curr. Opin. Pharmacol. 2006, 6, 468–472.
- Lei, J.; Sun, L.; Huang, S.; Zhu, C.; Li, P.; He, J.; Mackey, V.; Coy, D.H.; He, Q. The Antimicrobial Peptides and Their Potential Clinical Applications. Am. J. Transl. Res. 2019, 11, 3919–3931.
- Jaradat, D.M.M. Thirteen Decades of Peptide Synthesis: Key Developments in Solid Phase Peptide Synthesis and Amide Bond Formation Utilized in Peptide Ligation. Amino Acids 2018, 50, 39–68.
- González-Henríquez, C.M.; Sarabia-Vallejos, M.A.; Rodríguez-Hernández, J. Strategies to Fabricate Polypeptide-Based Structures via Ring-Opening Polymerization of N-Carboxyanhydrides. Polymers 2017, 9, 551.
- Cardoso, M.H.; Orozco, R.Q.; Rezende, S.B.; Rodrigues, G.; Oshiro, K.G.N.; Cândido, E.S.; Franco, O.L. Computer-Aided Design of Antimicrobial Peptides: Are We Generating Effective Drug Candidates? Front. Microbiol. 2020, 10, 3097.
- Mohamed, M.F.; Abdelkhalek, A.; Seleem, M.N. Evaluation of Short Synthetic Antimicrobial Peptides for Treatment of Drug-Resistant and Intracellular Staphylococcus Aureus. Sci. Rep. 2016, 6, 29707.
- Goodman, M.; Zapf, C.; Rew, Y. New Reagents, Reactions, and Peptidomimetics for Drug Design. Biopolymers 2001, 60, 229–245.
- Chou, K.C. Prediction of Protein Cellular Attributes Using Pseudo-Amino Acid Composition. Proteins 2001, 43, 246–255.
- Adessi, C.; Soto, C. Converting a Peptide into a Drug: Strategies to Improve Stability and Bioavailability. Curr. Med. Chem. 2002, 9, 963–978.
- Banerjee, A.; Pramanik, A.; Bhattacharjya, S.; Balaram, P. Omega Amino Acids in Peptide Design: Incorporation into Helices. Biopolymers 1996, 39, 769–777.
- Godballe, T.; Nilsson, L.L.; Petersen, P.D.; Jenssen, H. Antimicrobial β-Peptides and α-Peptoids. Chem. Biol. Drug Des. 2011, 77, 107–116.
- Evans, B.J.; King, A.T.; Katsifis, A.; Matesic, L.; Jamie, J.F. Methods to Enhance the Metabolic Stability of Peptide-Based PET Radiopharmaceuticals. Molecules 2020, 25, 2314.
- Gan, B.H.; Gaynord, J.; Rowe, S.M.; Deingruber, T.; Spring, D.R. The Multifaceted Nature of Antimicrobial Peptides: Current Synthetic Chemistry Approaches and Future Directions. Chem. Soc. Rev. 2021, 50, 7820–7880.
- Falciani, C.; Lozzi, L.; Scali, S.; Brunetti, J.; Bracci, L.; Pini, A. Site-Specific Pegylation of an Antimicrobial Peptide Increases Resistance to Pseudomonas Aeruginosa Elastase. Amino Acids 2014, 46, 1403–1407.
- Gong, Y.; Andina, D.; Nahar, S.; Leroux, J.-C.; Gauthier, M.A. Releasable and Traceless PEGylation of Arginine-Rich Antimicrobial Peptides. Chem. Sci. 2017, 8, 4082–4086.
- Rounds, T.; Straus, S.K. Lipidation of Antimicrobial Peptides as a Design Strategy for Future Alternatives to Antibiotics. Int. J. Mol. Sci. 2020, 21, 9692.
- Tang, J.; Chen, H.; He, Y.; Sheng, W.; Bai, Q.; Wang, H. Peptide-Guided Functionalization and Macrocyclization of Bioactive Peptidosulfonamides by Pd(II)-Catalyzed Late-Stage C-H Activation. Nat. Commun. 2018, 9, 3383.
- Abbasi, E.; Aval, S.F.; Akbarzadeh, A.; Milani, M.; Nasrabadi, H.T.; Joo, S.W.; Hanifehpour, Y.; Nejati-Koshki, K.; Pashaei-Asl, R. Dendrimers: Synthesis, Applications, and Properties. Nanoscale Res. Lett. 2014, 9, 247.
- Tam, J.P. Synthetic Peptide Vaccine Design: Synthesis and Properties of a High-Density Multiple Antigenic Peptide System. Proc. Natl. Acad. Sci. USA 1988, 85, 5409–5413.
- Tam, J.P.; Spetzler, J.C. Synthesis and Application of Peptide Dendrimers as Protein Mimetics. Curr. Protoc. Immunol. 2001, 9, 96.
- Bracci, L.; Falciani, C.; Lelli, B.; Lozzi, L.; Runci, Y.; Pini, A.; De Montis, M.G.; Tagliamonte, A.; Neri, P. Synthetic Peptides in the Form of Dendrimers Become Resistant to Protease Activity. J. Biol. Chem. 2003, 278, 46590–46595.
- Falciani, C.; Lozzi, L.; Pini, A.; Corti, F.; Fabbrini, M.; Bernini, A.; Lelli, B.; Niccolai, N.; Bracci, L. Molecular Basis of Branched Peptides Resistance to Enzyme Proteolysis. Chem. Biol. Drug Des. 2007, 69, 216–221.
- Wadhwani, P.; Reichert, J.; Bürck, J.; Ulrich, A.S. Antimicrobial and Cell-Penetrating Peptides Induce Lipid Vesicle Fusion by Folding and Aggregation. Eur. Biophys. J. 2012, 41, 177–187.
- Dong, N.; Chou, S.; Li, J.; Xue, C.; Li, X.; Cheng, B.; Shan, A.; Xu, L. Short Symmetric-End Antimicrobial Peptides Centered on β-Turn Amino Acids Unit Improve Selectivity and Stability. Front. Microbiol. 2018, 9, 2832.
- Yeaman, M.R.; Yount, N.Y. Mechanisms of Antimicrobial Peptide Action and Resistance. Pharmacol. Rev. 2003, 55, 27–55.
- McHenry, A.J.; Sciacca, M.F.M.; Brender, J.R.; Ramamoorthy, A. Does Cholesterol Suppress the Antimicrobial Peptide Induced Disruption of Lipid Raft Containing Membranes? Biochim. Biophys. Acta 2012, 1818, 3019–3024.
- Silhavy, T.J.; Kahne, D.; Walker, S. The Bacterial Cell Envelope. Cold Spring Harb. Perspect. Biol. 2010, 2, a000414.
- Malanovic, N.; Lohner, K. Antimicrobial Peptides Targeting Gram-Positive Bacteria. Pharmaceuticals 2016, 9, 59.
- Li, J.; Koh, J.-J.; Liu, S.; Lakshminarayanan, R.; Verma, C.S.; Beuerman, R.W. Membrane Active Antimicrobial Peptides: Translating Mechanistic Insights to Design. Front. Neurosci. 2017, 11, 73.
- Wimley, W.C. Describing the Mechanism of Antimicrobial Peptide Action with the Interfacial Activity Model. ACS Chem. Biol. 2010, 5, 905–917.
- Brogden, K.A. Antimicrobial Peptides: Pore Formers or Metabolic Inhibitors in Bacteria? Nat. Rev. Microbiol. 2005, 3, 238–250.
- Manzini, M.C.; Perez, K.R.; Riske, K.A.; Bozelli, J.C.; Santos, T.L.; da Silva, M.A.; Saraiva, G.K.V.; Politi, M.J.; Valente, A.P.; Almeida, F.C.L.; et al. Peptide:Lipid Ratio and Membrane Surface Charge Determine the Mechanism of Action of the Antimicrobial Peptide BP100. Conformational and Functional Studies. Biochim. Biophys. Acta 2014, 1838, 1985–1999.
- Yang, L.; Harroun, T.A.; Weiss, T.M.; Ding, L.; Huang, H.W. Barrel-Stave Model or Toroidal Model? A Case Study on Melittin Pores. Biophys. J. 2001, 81, 1475–1485.
- Kumar, S.; Singh, D.; Kumari, P.; Malik, R.S.; Poonam, P.K.; Parang, K.; Tiwari, R.K. PEGylation and Cell-Penetrating Peptides: Glimpse into the Past and Prospects in the Future. Curr. Top. Med. Chem. 2020, 20, 337–348.
- Sengupta, D.; Leontiadou, H.; Mark, A.E.; Marrink, S.-J. Toroidal Pores Formed by Antimicrobial Peptides Show Significant Disorder. Biochim. Biophys. Acta 2008, 1778, 2308–2317.
- Tuerkova, A.; Kabelka, I.; Králová, T.; Sukeník, L.; Pokorná, Š.; Hof, M.; Vácha, R. Effect of Helical Kink in Antimicrobial Peptides on Membrane Pore Formation. eLife 2020, 9, e47946.
- Clark, S.; Jowitt, T.A.; Harris, L.K.; Knight, C.G.; Dobson, C.B. The Lexicon of Antimicrobial Peptides: A Complete Set of Arginine and Tryptophan Sequences. Commun. Biol. 2021, 4, 605.
- Moravej, H.; Moravej, Z.; Yazdanparast, M.; Heiat, M.; Mirhosseini, A.; Moosazadeh Moghaddam, M.; Mirnejad, R. Antimicrobial Peptides: Features, Action, and Their Resistance Mechanisms in Bacteria. Microb. Drug Resist. 2018, 24, 747–767.
- Le, C.-F.; Fang, C.-M.; Sekaran, S.D. Intracellular Targeting Mechanisms by Antimicrobial Peptides. Antimicrob. Agents Chemother. 2017, 61, e02340–16.
- Birkemo, G.A.; Lüders, T.; Andersen, Ø.; Nes, I.F.; Nissen-Meyer, J. Hipposin, a Histone-Derived Antimicrobial Peptide in Atlantic Halibut (Hippoglossus Hippoglossus L.). Biochim. Biophys. Acta 2003, 1646, 207–215.
- Marchand, C.; Krajewski, K.; Lee, H.-F.; Antony, S.; Johnson, A.A.; Amin, R.; Roller, P.; Kvaratskhelia, M.; Pommier, Y. Covalent Binding of the Natural Antimicrobial Peptide Indolicidin to DNA Abasic Sites. Nucleic Acids Res. 2006, 34, 5157–5165.
- Braffman, N.R.; Piscotta, F.J.; Hauver, J.; Campbell, E.A.; Link, A.J.; Darst, S.A. Structural Mechanism of Transcription Inhibition by Lasso Peptides Microcin J25 and Capistruin. Proc. Natl. Acad. Sci. USA 2019, 116, 1273–1278.
- Heddle, J.G.; Blance, S.J.; Zamble, D.B.; Hollfelder, F.; Miller, D.A.; Wentzell, L.M.; Walsh, C.T.; Maxwell, A. The Antibiotic Microcin B17 Is a DNA Gyrase Poison: Characterisation of the Mode of Inhibition11Edited by J. Karn. J. Mol. Biol. 2001, 307, 1223–1234.
- Polikanov, Y.S.; Aleksashin, N.A.; Beckert, B.; Wilson, D.N. The Mechanisms of Action of Ribosome-Targeting Peptide Antibiotics. Front. Mol. Biosci. 2018, 5, 48.
- Friedrich, C.L.; Rozek, A.; Patrzykat, A.; Hancock, R.E.W. Structure and Mechanism of Action of an Indolicidin Peptide Derivative with Improved Activity against Gram-Positive Bacteria*. J. Biol. Chem. 2001, 276, 24015–24022.
- Patrzykat, A.; Friedrich, C.L.; Zhang, L.; Mendoza, V.; Hancock, R.E.W. Sublethal Concentrations of Pleurocidin-Derived Antimicrobial Peptides Inhibit Macromolecular Synthesis in Escherichia Coli. Antimicrob. Agents Chemother. 2002, 46, 605–614.
- Ho, Y.-H.; Shah, P.; Chen, Y.-W.; Chen, C.-S. Systematic Analysis of Intracellular-Targeting Antimicrobial Peptides, Bactenecin 7, Hybrid of Pleurocidin and Dermaseptin, Proline–Arginine-Rich Peptide, and Lactoferricin B, by Using Escherichia Coli Proteome Microarrays*. Mol. Cell. Proteom. 2016, 15, 1837–1847.
- Florin, T.; Maracci, C.; Graf, M.; Karki, P.; Klepacki, D.; Berninghausen, O.; Beckmann, R.; Vázquez-Laslop, N.; Wilson, D.N.; Rodnina, M.V.; et al. An Antimicrobial Peptide That Inhibits Translation by Trapping Release Factors on the Ribosome. Nat. Struct. Mol. Biol. 2017, 24, 752–757.
- El-Mowafi, S.A.; Sineva, E.; Alumasa, J.N.; Nicoloff, H.; Tomsho, J.W.; Ades, S.E.; Keiler, K.C. Identification of Inhibitors of a Bacterial Sigma Factor Using a New High-Throughput Screening Assay. Antimicrob. Agents Chemother. 2015, 59, 193–205.
- Van Eijk, E.; Wittekoek, B.; Kuijper, E.J.; Smits, W.K. DNA Replication Proteins as Potential Targets for Antimicrobials in Drug-Resistant Bacterial Pathogens. J. Antimicrob. Chemother. 2017, 72, 1275–1284.
- Knappe, D.; Goldbach, T.; Hatfield, M.P.D.; Palermo, N.Y.; Weinert, S.; Sträter, N.; Hoffmann, R.; Lovas, S. Proline-Rich Antimicrobial Peptides Optimized for Binding to Escherichia Coli Chaperone DnaK. Protein Pept. Lett. 2016, 23, 1061–1071.
- Bhowmick, M.; Tokmina-Roszyk, D.; Onwuha-Ekpete, L.; Harmon, K.; Robichaud, T.; Fuerst, R.; Stawikowska, R.; Steffensen, B.; Roush, W.; Wong, H.R.; et al. Second Generation Triple-Helical Peptide Inhibitors of Matrix Metalloproteinases. J. Med. Chem. 2017, 60, 3814–3827.
- Tay, C.X.; Quah, S.Y.; Lui, J.N.; Yu, V.S.H.; Tan, K.S. Matrix Metalloproteinase Inhibitor as an Antimicrobial Agent to Eradicate Enterococcus Faecalis Biofilm. J. Endod. 2015, 41, 858–863.
- Rahman, F.; Nguyen, T.-M.; Adekoya, O.A.; Campestre, C.; Tortorella, P.; Sylte, I.; Winberg, J.-O. Inhibition of Bacterial and Human Zinc-Metalloproteases by Bisphosphonate- and Catechol-Containing Compounds. J. Enzyme Inhib. Med. Chem. 2021, 36, 819–830.
- Salomón, R.A.; Farías, R.N. Microcin 25, a Novel Antimicrobial Peptide Produced by Escherichia Coli. J. Bacteriol. 1992, 174, 7428–7435.
- Chileveru, H.R.; Lim, S.A.; Chairatana, P.; Wommack, A.J.; Chiang, I.-L.; Nolan, E.M. Visualizing Attack of Escherichia Coli by the Antimicrobial Peptide Human Defensin 5. Biochemistry 2015, 54, 1767–1777.
- Scherer, K.M.; Spille, J.-H.; Sahl, H.-G.; Grein, F.; Kubitscheck, U. The Lantibiotic Nisin Induces Lipid II Aggregation, Causing Membrane Instability and Vesicle Budding. Biophys. J. 2015, 108, 1114–1124.
- Wiedemann, I.; Böttiger, T.; Bonelli, R.R.; Schneider, T.; Sahl, H.-G.; Martínez, B. Lipid II-Based Antimicrobial Activity of the Lantibiotic Plantaricin C. Appl. Environ. Microbiol. 2006, 72, 2809–2814.
- Brötz, H.; Bierbaum, G.; Reynolds, P.E.; Sahl, H.-G. The Lantibiotic Mersacidin Inhibits Peptidoglycan Biosynthesis at the Level of Transglycosylation. Eur. J. Biochem. 1997, 246, 193–199.
- Shanmugaraj, B.; Bulaon, C.J.I.; Malla, A.; Phoolcharoen, W. Biotechnological Insights on the Expression and Production of Antimicrobial Peptides in Plants. Molecules 2021, 26, 4032.
- Rezende, S.B.; Oshiro, K.G.N.; Júnior, N.G.O.; Franco, O.L.; Cardoso, M.H. Advances on Chemically Modified Antimicrobial Peptides for Generating Peptide Antibiotics. Chem. Commun. 2021, 57, 3793.
- Batoni, G.; Maisetta, G.; Esin, S. Antimicrobial Peptides and Their Interaction with Biofilms of Medically Relevant Bacteria. Biochim. Biophys. Acta 2016, 1858, 1044–1060.
- Wei, H.; Xie, Z.; Tan, X.; Guo, R.; Song, Y.; Xie, X.; Wang, R.; Li, L.; Wang, M.; Zhang, Y. Temporin-Like Peptides Show Antimicrobial and Anti-Biofilm Activities against Streptococcus Mutans with Reduced Hemolysis. Molecules 2020, 25, 5724.
- Rinaldi, A.C.; Conlon, J.M. The Temporins. In Hanbook of Biologically Active Peptides; Kastin, A.J., Ed.; Elsevier: San Diego, CA, USA, 2013; pp. 400–406.
- Nuri, R.; Shprung, T.; Shai, Y. Defensive Remodeling: How Bacterial Surface Properties and Biofilm Formation Promote Resistance to Antimicrobial Peptides. Biochim. Biophys. Acta 2015, 1848, 3089–3100.
- Band, V.I.; Weiss, D.S. Mechanisms of Antimicrobial Peptide Resistance in Gram-Negative Bacteria. Antibiotics 2015, 4, 18–41.
- Joo, H.-S.; Fu, C.-I.; Otto, M. Bacterial Strategies of Resistance to Antimicrobial Peptides. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2016, 371, 20150292.
- Magana, M.; Pushpanathan, M.; Santos, A.L.; Leanse, L.; Fernandez, M.; Ioannidis, A.; Giulianotti, M.A.; Apidianakis, Y.; Bradfute, S.; Ferguson, A.L.; et al. The Value of Antimicrobial Peptides in the Age of Resistance. Lancet Infect. Dis. 2020, 20, e216–e230.
- Annunziato, G.; Costantino, G. Antimicrobial Peptides (AMPs): A Patent Review (2015–2020). Expert Opin. Ther. Pat. 2020, 30, 931–947.
- Guan, Q.; Huang, S.; Jin, Y.; Campagne, R.; Alezra, V.; Wan, Y. Recent Advances in the Exploration of Therapeutic Analogues of Gramicidin S, an Old but Still Potent Antimicrobial Peptide. J. Med. Chem. 2019, 62, 7603–7617.
- Rodríguez-Santiago, J.; Cornejo-Juárez, P.; Silva-Sánchez, J.; Garza-Ramos, U. Polymyxin Resistance in Enterobacterales: Overview and Epidemiology in the Americas. Int. J. Antimicrob. Agents 2021, 16, 106426.
- Gogry, F.A.; Siddiqui, M.T.; Sultan, I.; Haq, Q.M.R. Current Update on Intrinsic and Acquired Colistin Resistance Mechanisms in Bacteria. Front. Med. 2021, 8, 677720.
- Huang, H.W. DAPTOMYCIN, Its Membrane-Active Mechanism vs. That of Other Antimicrobial Peptides. Biochim. Biophys. Acta Biomembr. 2020, 1862, 183395.
- Liu, W.-T.; Chen, E.-Z.; Yang, L.; Peng, C.; Wang, Q.; Xu, Z.; Chen, D.-Q. Emerging Resistance Mechanisms for 4 Types of Common Anti-MRSA Antibiotics in Staphylococcus Aureus: A Comprehensive Review. Microb. Pathog. 2021, 156, 104915.