Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Beatrix Zheng and Version 1 by Maria De la Fuente.
The development and commercialization of mRNA COVID-19 vaccines have pivoted the way towards future applications of mRNA medicines by finally finding a solution to the problem of delivery. Despite concerns from detractors about a lack of applications for nanotechnology, nanomedicine has now demonstrated that it is a translational, relevant and much-needed approach to engineer new genetic drugs. Thus, it is now clear that the full potential of mRNA therapeutics, along with its advantages could not be envisioned without nanomedicine.
- mRNA
- LNPs
- vaccine
- COVID-19
- nanomedicine
- nanotechnology
- gene therapy
- CRISPR
- cancer
- protein replacement
1. Prospects and Limitations of mRNA
The first isolation of messenger RNA (mRNA) was published in Nature in 1961 [1[1][2],2], starting the path towards a full understanding of this molecule [3]. The potential to synthetically produce mRNA from DNA in the 1980s, i.e., in vitro transcribed mRNA (IVT mRNA), opened a broad spectrum of preclinical applications [4]. Since then, multiple efforts have been performed to understand its mechanism of action and the path towards developing mRNA-based drugs. Therapeutic mRNA’s potential lies in its capacity to induce the expression of proteins [4,5][4][5] for preventing or altering a particular disease state. mRNA therapeutics hold many opportunities, which can depend on the targeted cells, organ selective accumulation and encoded protein of interest. Nowadays, however, two main approaches are considered when using mRNA, both of which will be reviewed and explained in depth. The first is dendritic cell (DC) targeting, so as to achieve immune activation. The second is to exploit the natural capacity of nanoparticles to accumulate in the liver and use it to endogenously produce therapeutic proteins.
Importantly, mRNA has several advantages over other gene therapies such as DNA or pDNA which make this molecule more translational in terms of pharmaceutical properties: (i) mRNA does not need to reach the nucleus of the cell as opposed to DNA, resulting in more efficient delivery; (ii) mRNA does not integrate into the genome of the host cell, a fact associated with risk of mutagenesis; (iii) mRNA can be synthesized in the lab with fairly easy protocols, following scalable processes in agreement with GMP regulations; and (iv) mRNA sequences can be easily modified and updated, which is an important fact to consider in vaccinology (i.e., when mutations of the target protein occur). These advantages are also important when comparing mRNA technology to protein delivery technology, which normally comes with short half-lives and expensive and tedious industrial processes [4,5,6][4][5][6].
2. Biomedical Applications of mRNA Using Nanomedicine
The approval of mRNA vaccines using LNPs can indeed be considered the biggest milestone to foster the clinical translation of many other mRNA nanoformulations that can address current clinical needs. Indeed, the technology can be applied to a broad spectrum of applications and therapeutic areas. Vaccines for infectious diseases might be the first application that has reached the market, but nanoformulated mRNA has the full potential to answer clinical demands related to the needs of therapeutic proteins. In the next sections, we aim to provide a full overview of the most promising developments to date. Biomedical applications for mRNA therapeutics are pictured in Figure 41.
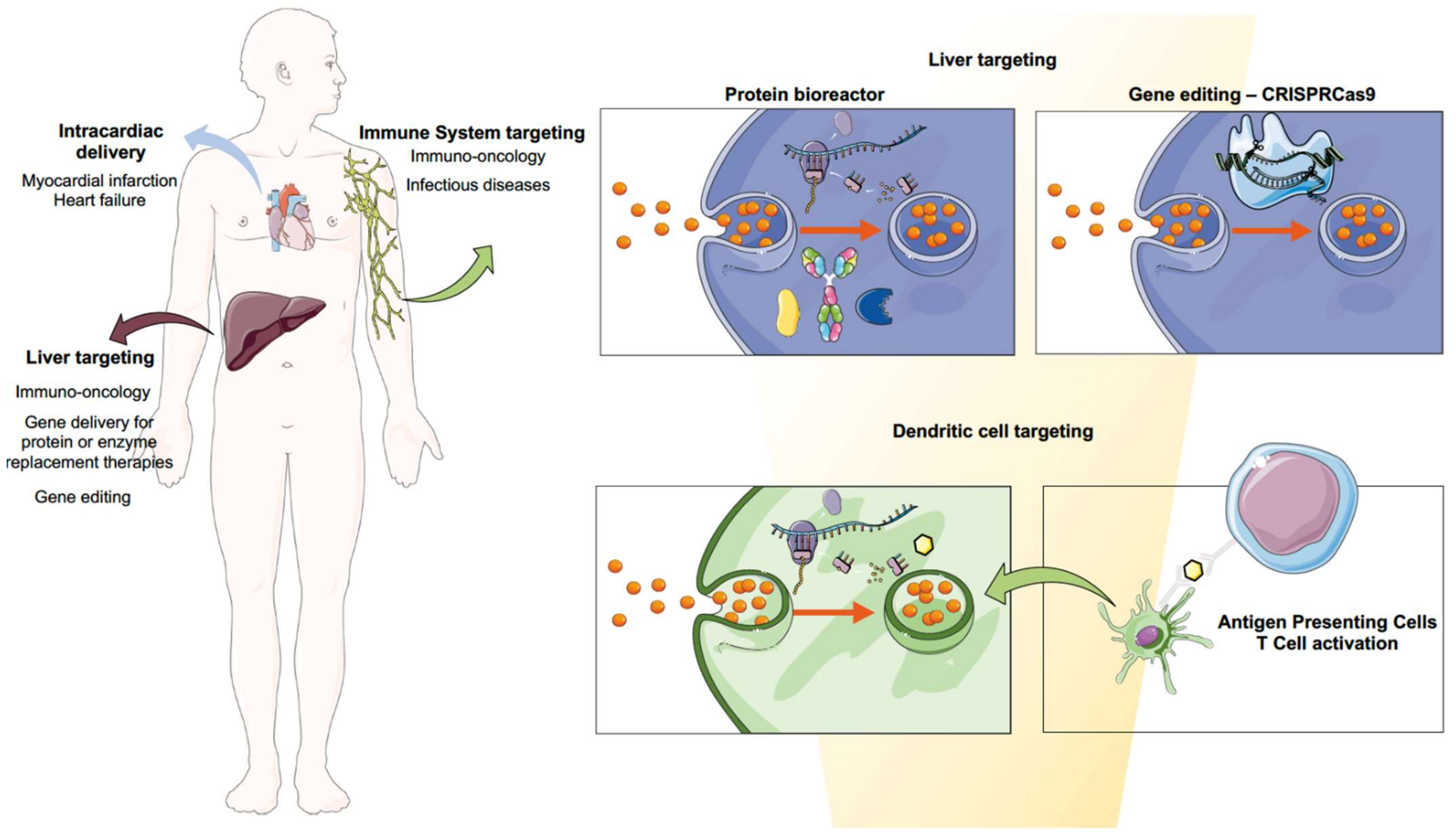
Figure 41. Biomedical applications for mRNA therapeutics. Immuno-oncology treatments can be envisioned using the liver as a factory of therapeutic proteins such as neoantigens or antibodies, as well as by using the antigen-presenting cells’ physiological function to induce immune activation towards cancerous cells. The latter can also be applied for the development of safe and efficacious mRNA and saRNA vaccines against infectious diseases. Gene delivery for therapeutic protein synthesis or restoration of a particular enzymatic function as well as gene editing using CRISPR cas9 technology can be achieved by targeting the liver. Cardiovascular regeneration can be achieved by intracardial delivery of mRNA encoding for growth factors.
2.1. Infectious Diseases
Despite the fact that COVID-19 vaccines have been the first marketed mRNA formulations, this technology has been researched for decades using different delivery approaches, from LPX to cationic nanoemulsions to polymeric nanoparticles [9][7]. Since mRNA loaded into liposomes demonstrated for the first time that it could trigger a specific T-cell response against influenza virus in mice [141][8], the field has enormously advanced. In fact, only in 2019, 15 mRNA candidates entered clinical trials [10][9]. Because this area has very recently been covered in an extensive review [142][10], wthe researchers will only showcase some of the most promising approaches, redirecting the reader to this article for further information. Nonetheless, all the clinical trials for infectious diseases other than COVID-19 are shown in Table 1. In this regard, clinically, respiratory syncytial virus (RSV) and influenza virus are amongst the most advanced clinical candidates.
Table 1. mRNA Nanomedicines for Infectious Diseases in Clinical Trials. A list of clinical trials for infectious diseases other than COVID-19 is provided in this table.
| Infectious Diseases Other than COVID-19 | |||||
|---|---|---|---|---|---|
| Therapeutic | Clinical Application | Formulation | Phase | NCT Number | Sponsor |
Today, both strategies described, (1) and (2), are under clinical evaluation in phase 1 or 2. Melanoma, prostate cancer, triple-negative breast cancer and head and neck cancer are some of the biomedical applications in which this technology is being clinically investigated (Table 2). An extensive review on this matter has been recently published [161][31].
Table 2. mRNA Nanomedicines for Oncology in Clinical Trials. A list of clinical trials for oncology applications, including the subsections described above, is provided in this table.
| Oncology | |||||
| Cancer Vaccines | |||||
| Shared Tumor Antigens | |||||
| BNT111 | Advanced Melanoma | Modified mRNA-LPX | Phase II | NCT04526899 | BioNTech |
| BNT112 | Prostate Cancer | Modified mRNA-LPX | Phase I/II | NCT04382898 | BioNTech |
| BNT113 | HPV16+ head and neck cancer | Modified mRNA-LPX | Phase II | NCT04534205 | BioNTech |
| BNT114 | Triple-Negative Breast Cancer | Modified mRNA-LPX | Phase I | NCT03815058 | BioNTech |
| BNT115 | Ovarian Cancer | Modified mRNA-LPX | Phase I | NCT04163094 | BioNTech |
| mRNA-5671 | Carcinoma, NSCLC, Pancreatic and colorectal neoplasms | Non-disclosed | Phase I | NCT03948763 | Merck/Moderna |
| Personalized cancer vaccines | |||||
| mRNA-4157 | Melanoma | Modified mRNA-LNPs | Phase II | NCT03897881 | Moderna |
| BNT122 | Advanced Melanoma | Modified mRNA-LPX | Phase I | NCT03815058 | BioNTech |
| mRNA-4157 | Solid Tumors | Modified mRNA-LNPs | Phase I | NCT03313778 | Moderna |
| mRNA-2752 | Solid Tumors | Modified mRNA-LNPs | Phase I | NCT03739931 | Moderna |
| CV8102 | Solid Tumors | Not disclosed | Phase I | NCT03291002 | CureVac |
| V941 | Solid Tumors | Not disclosed | Phase I | NCT03948763 | Merck Sharp & Dohme Corp. |
| Medi1191 | Solid Tumors | Modified mRNA-LPNs | Phase I | NCT03946800 | MedImmuneLC |
| mRNA-2416 | Solid Tumors and Lymphoma | Modified mRNA-LNPs | Phase I/II | NCT03323398 | Moderna |
| mRNA-2752 | Solid Tumors and Lymphoma | Modified mRNA-LNPs | Phase I | NCT03739931 | Moderna |
| mRNA-2752 | Carcinoma | Modified mRNA-LNPs | Phase I | NCT02872025 | Moderna |
| BNT122 | Colorectal Cancer Stage II/III | Modified mRNA-LPX | Phase II | NCT04486378 | BioNTech |
| MVT-5873 | Pancreatic Cancer | Not disclosed | Phase I | NCT02672917 | BioNTech |
| BNT131 | Metastatic Neoplasm | Not disclosed | Phase I | NCT03871348 | BioNTech |
| The Liver as a Bioreactor | |||||
| Encoding Cytokines | |||||
| BNT152, BNT153 | Solid Tumor | Not disclosed | Phase I | NCT04710043 | BioNTech |
| BNT151 | Solid Tumor | Not disclosed | Phase I/II | NCT04455620 | BioNTech |
| mRNA-6231 | Autoimmune Disorders | Modified mRNA-LNPs | Phase I | NCT04916431 | Moderna |
| Encoding Antibodies | |||||
| BNT142 | Solid Tumor | Encoding Ab | Phase I/II | NCT04503278 | BioNTech |
| mRNA-1944 | Chikungunya | Modified mRNA-LNPs | Phase I | NCT03829384 | Moderna |
| mRNA-1653 | hMPV/PIV3 | Modified mRNA-LNPs | Phase I | NCT04144348 NCT03392389 |
Moderna |
| mRNA-1345 | RSV | Modified mRNA-LNPs | Phase I | NCT04528719 | Moderna |
| mRNA-11777 | RSV | Modified mRNA-LNPs | Phase I | Unregistered | Moderna, Merck |
| mRNA-1172 | RSV | Modified mRNA-LNPs | Phase I | Unregistered | Moderna, Merck |
| mRNA-1893 | Zika | Modified mRNA-LNPs | Phase I | NCT04064905 | Moderna |
| mRNA-1325 | Zika | Modified mRNA-LNPs | Phase I | NCT03014089 | Moderna |
| mRNA-1647 | CMV | Modified mRNA-LNPs | Phase II | NCT04232280 | Moderna |
| mRNA-1647 | CMV | Modified mRNA-LNPs | Phase II | NCT03382405 | Moderna |
| mRNA-1443 | CMV | Modified mRNA-LNPs | Phase I | NCT03382405 | Moderna |
| MRT5400 | Influenza A (H3N2) | Not disclosed | Phase I | Unregistered | TranslateBio, Sanofi |
| MRT5401 | Influenza A (H3N2) | Not disclosed | Phase I | Unregistered | TranslateBio, Sanofi |
| mRNA-1010 | Influenza A, Influenza B | Not disclosed | Phase II/III | NCT04956575 | Moderna |
| mRNA-1851 | Influenza A (H7N9) | Modified mRNA-LNPs | Phase I | NCT03345043 | Moderna |
| mRNA-1440 | Influenza A (H7N8) | Modified mRNA-LNPs | Phase I | NCT03076385 | Moderna |
| mRNA-1388 | Chikungunya | Modified mRNA-LNPs | Phase I | NCT03829384 | Moderna |
| CV7201 | Rabies | Non-modified mRNA- in RNActive | Phase I | NCT02241135 | CureVac |
| CV7202 | Rabies | Unmodified mRNA-LNPs | Phase I | NCT03713086 | CureVac |
| GSK3903133A | Rabies | saRNA–cationic nanoemulsion | Phase I | NCT04062669 | GSK |
Abbreviations: hMPV/PIV3—human metapneumovirus/parainfluenzavirus type 3; RSV—respiratory syncytial virus; CMV—cytomegalovirus; HPV—human papillomavirus.
Furthermore, preclinically, one of the most important applications is the HIV vaccine. HIV currently affects 38 million people; these numbers are expected to increase by 2030 [143][11]. This virus has been extensively researched for decades; however, its retroviral nature, along with the fact that it possesses a very high antigen diversity in its envelope, make it very difficult to achieve good outcomes [144][12]. In recent years, several studies have tried to deliver mRNA-vaccine encoding for HIV proteins; however, their success has not been as expected [127,129,145][13][14][15]. Nevertheless, a recently published study has shown broad neutralization and reduction in the risk of infection in rhesus macaques upon the administration of mRNA encoding for both HIV-1 envelope protein and simian immunodeficiency virus Gag proteins, which would be responsible for forming virus-like particles (VLP) [146][16]. A different approach based on targeting hepatocytes, which will be further discussed for immuno-oncology applications in Section 3.2.2below, has achieved good results. This sentudry involved the delivery of mRNA encoding the light and heavy chains of VRC01, a neutralizing antibody against HIV-1. Using LNPs, they were able to achieve robust protein expression in the liver. Furthermore, prophylactic immunization was also achieved at a dose of 0.7 mg/kg as opposed to the 10–20 mg/kg needed if the recombinant protein is administered [147][17].
Another important preclinical advancement was achieved with the Ebola vaccine. Ebola virus (EbolV) caused the deaths of more than 11,000 lives across West Africa from 2014 to 2016 [148][18]. This lethal virus causes a hemorrhagic fever that is fatal in up to 50% of the patients [148][18]. In 2019, one viral vector-based Ebola vaccine was approved by the FDA; however, clinical trials reported some safety concerns. As explained in the introduction, from this point of view, mRNA vaccination presents an advantage. In a study on guinea pigs, 20 μg of glycoprotein-encoding mRNA was administered using LNPs as a delivery vehicle, resulting in good antibody titers and protection from death [149][19].
Lastly, it is important to note the advancements achieved in other infectious diseases other than viruses. In this regard, Plasmodium, the causal agent of Malaria, is highlighted next. Very recently, the first malaria vaccine was approved by WHO [150][20]. The RTS, S malaria vaccine is a recombinant protein vaccine which has shown to prevent lethal disease, and is especially efficacious when given together with pharmacological treatment [150][20]. Several studies have also been conducted towards the development of an mRNA vaccine against malaria. The first is an saRNA-based [151][21] encoding for Plasmodium-secreted cytokine macrophage migrating inhibitory factor, which had previously shown efficacy [152][22]. In this study, anti-Plasmodium antibodies and protective T-cell memory were achieved [151][21]. In another study, Plasmodium falciparum acid-rich protein (PfGARP) mRNA-encoded LNPs were dosed to infected aotus monkeys, showing reduced levels of the parasite after three doses [153][23].
Altogether, these advancements provide strong evidence of the possibilities ahead regarding mRNA vaccinology, which seems to provide necessary advantages as the next vaccine technology, especially when considering future pandemics.
2.2. Immuno-Oncology
RNAs are critical for gene expression in their multiple forms, whether in non-coding forms such as miRNA, siRNA or tRNAs that regulate transcription and translation, or in coding forms such as mRNA [154][24]. Cancer immunotherapy has been a game changer since the approval of the immune checkpoint inhibitor ipilimumab in 2011 [155][25]. The number of immuno-oncology (IO) drugs in the development pipeline in 2020 grew by 22% compared to 2019, accentuating the high interest in these therapies. Immunomodulators, cell therapies and cancer vaccines represent the most important trends [156][26]. In fact, these therapies are often given simultaneously, with one example of this being TriMiX, whose composition is CD70, CD40 ligand and TLR4 [157][27]. However, scientists have met a few challenges when it comes to the new development of these immunotherapeutic agents, mostly related to off-target toxicity [158][28]. Thus, mRNA technology can offer advantages in terms of scalability, cost and production issues, widening immunotherapy use worldwide. Altogether, immunotherapy combined with nanomedicine could bring a potential solution: to engineer the delivery of immunomodulators, focusing their action on target tissues such as tumors or tumor-draining lymph nodes, or specific cell types, so as to control the timing and location of immunomodulation [158][28]. To achieve this, two main strategies can be performed: the first, by targeting DCs and taking advantage of their natural immune role; and the second, by making use of liver capacity to synthesize proteins. In the following sections we will describe both mechanisms in depth.
2.2.1. Cancer Vaccines: Targeting Dendritic Cells
The most well-known strategy for treating cancer with mRNA is cancer vaccines, in which mRNA is transcribed into a protein which in turn trains the immune system to fight against the tumor. Thus, rather than being prophylactic, cancer vaccines seek to stimulate cell-mediated responses. Therapeutically, this technology can be applied (1) by using shared antigens for the same tumor to stimulate immune activation, or (2) by designing a personalized cancer vaccine which possesses the individual’s neoantigens, i.e., proteins preferentially expressed in cancerous cells but not in the patients’ healthy tissue, such as growth-associated factors or antigens [9][7].
The way the latter strategy (2) is approached involves the most advanced tissue gene sequencing: a patients’ tumor and healthy tissue samples are taken and analyzed looking for tumor-associated antigens, as pictured in Figure 52.
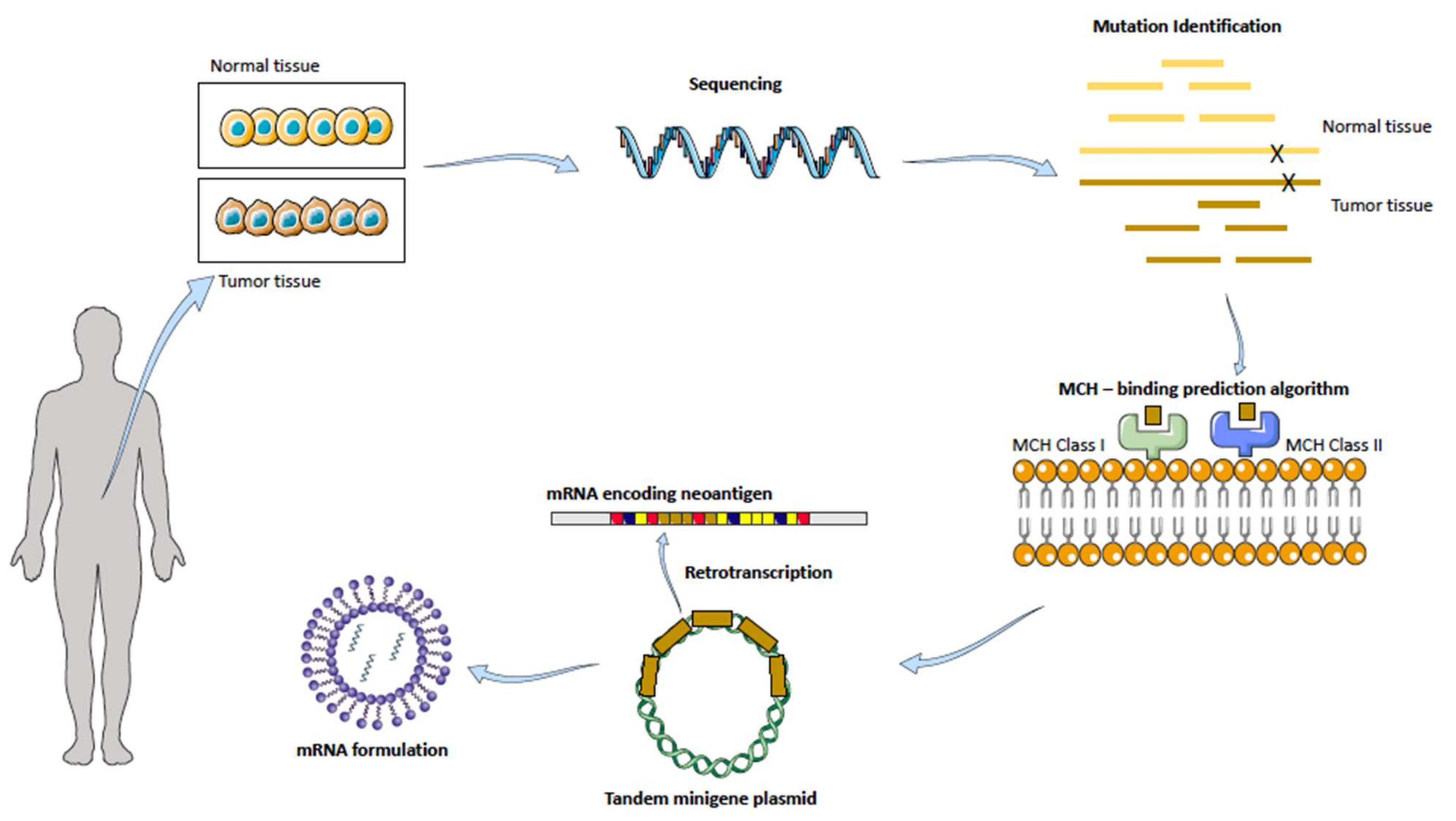
Figure 52. Personalized mRNA cancer vaccines. Healthy and tumor tissue are isolated from the patient sample, analyzed and compared in order to find mutations present in the tumor tissue. Bioinformatic tools are used to predict the MCH Class I and II binding. A tandem gene encoding for several neoantigens is then designed and retrotranscribed to mRNA. Finally, by making use of nanotechnology, the mRNA is formulated and delivered. Adapted with permission from [159][29], published by Nature Reviews Drug Discovery, 2018.
The technology relies on a special algorithm prediction of binding strength to self-major histocompatibility complex (MHC) receptors. After mutation identification, neoantigens are ranked based on how well they would bind to this receptor, which will then drive the activation of T cells, followed by the immunological response against that specific neoantigen. Upon administration of the mRNA into a suitable, stable and efficient formulation, DCs are transfected, and due to their antigen-presenting cells’ nature, they localize the neoantigen onto their MHC receptor located in their surface, which will be then presented to the T cells, starting the immune cascade [159,160][29
Abbreviations: Ab—antibodies; NSCLC—non-small-cell lung cancer.
2.2.2. The Liver as a Factory of Immunomodulatory Proteins
As commented in the previous sectionly, targeting DCs is the main strategy towards the development of cancer vaccines. Nonetheless, this is not the only approach in which mRNA can be used for immunotherapy. This approach is based on the on the fact that nanoparticles have the intrinsic capacity to accumulate in the liver [162][32], which has been for many years the core center of the research. Thus, targeting the liver can be achieved by exploiting its capacity for protein production in order to build up an endogenous factory of therapeutic proteins, which will then be released into systemic circulation. In fact, the feasibility of targeting the liver for mRNA delivery for the endogenous production of immunomodulators can be envisioned by considering advances in other research areas, such as protein replacement therapies with mRNA, which will also be covered in Section 3.3below. Furthermore, this strategy also overcomes the main disadvantages of many immunomodulator administrations, such as their short half-life and subsequent long perfusions and recurrent visits to the medical doctor’s office, as well as their high production cost [163][33].
This mRNA application has been studied for two types of recombinant proteins: immunomodulators and antibodies. The production of immunomodulators, such as cytokines, has already been proven to be an achievable strategy. For instance, intratumoral administration of LNPs with mRNA-encoding cytokines IL-23 and IL-36 and the T-cell costimulatory OX40L was proven to lead to tumor regression in colon cancer and melanoma models and is now under clinical trials (NCT03739931) [164][34]. In another study using IL-12, a well-known cytokine [165][35], Lai et al. reported mRNA-encoding IL-12 LNPs to be a good candidate for slowing down the progression of MYC oncogene-driven human hepatocellular carcinoma, with good biodistribution within the tumor and no toxicity. Treatment reduced liver-tumor burden and increased survival in MYC-induced HCC transgenic mice [166][36].
Interestingly, co-delivery of cytokines and antibodies has proven to have potential for treating different types of cancer [166,167,168][36][37][38]. In fact, the use of mRNA to endogenously produce antibodies has also been achieved. Apart from the already-commented HIV study inbefore Section 3.1 [147][17], another study with trastuzumab aimed to apply this strategy to tackle oncology. Rybakova et al. succeeded in efficiently delivering mRNA encoding for trastuzumab, a monoclonal antibody widely used for breast cancer. They found a dose-dependent increase in trastuzumab protein levels in mouse serum after 24 h administration. When the administration of the monoclonal antibody Herceptin® was compared, results revealed similar half-lives for both antibodies (i.e., endogenously produced and Herceptin); however, the mRNA-based delivery resulted in a 64% higher antibody serum concentration over the course of 30 days. Overall, a single dose of LNPs with the mRNA at 2 mg/kg showed a favorable pharmacokinetic profile when compared to a single injection of Herceptin at 8 mg/kg [169][39]. This technology has also been proven to be efficient for the delivery of bispecific antibodies [170][40]. Stadler et al. succeeded in achieving a promising therapeutic outcome by generating his-tagged bispecific antibodies directed against the T-cell receptor-associated molecule CD3, a tumor-associated antigen [170][40].
2.2.3. CAR-T Cells
On a last note on immunotherapy, the CAR-T-cell field can also take advantage of mRNA technology. Chimeric antigen receptor (CAR) T-cell therapy is considered by many the next promise of cancer immunotherapy. In August 2017, the FDA approved the first CAR-T treatment, Kymriah® (tisagenlecleucel), for the treatment of certain patients, both pediatric and young adults with B-cell acute lymphoblastic leukemia (B-ALL). Several months after that, in October 2017, the FDA approved Yescarta® (axicabtagene ciloleucel) for adult patients with certain types of refractory B-cell non-Hodgkin lymphoma (B-NHL) [171][41] This approach consists of engineering lymphocyte T surfaces with tumor specific antigens that would bind to tumor-specific receptors. Right now, CD19-targeting CAR-T-cell therapy is approved for both B-cell acute lymphoblastic leukemia (B-ALL) as well as non-Hodgkin lymphoma (NHL), where it has demonstrated up to 90% complete remission when rayed in chemo-refractory B-cell malignancies [171][41]. Despite the promising positive outcomes on liquid tumors, CAR-T therapy has challenges in its application to solid tumors, including limited cancer-specific targets and non-persistence of the transferred CAR-T cells, allowing for low side effects but efficient tumor eradication [172][42], and research needs to be carried out to tackle solid tumors such as breast, melanoma or ovarian. With regard to solid tumors, in a recent paper published, T-cells were bioengineered with nanoparticle-encapsulating mRNA encoding for a single-chain variable fragment (scFv) that can specifically bind to CLDN6, an oncofetal cell-surface antigen for CAR-T-cell targeting. Results demonstrated that RNA nanoparticles can efficiently stimulate CAR-T cells [173][43].
2.3. RNA-Based Protein Replacement Therapies
As mentioned before, attempts to deliver therapeutic antibodies as mRNA molecules have been carried out with success with the aim of overcoming the drawbacks of administering the therapeutic protein. So far, protein replacement therapies are mainly focused on secreted proteins or proteins of bacterial origin. It is important to understand the processes following the translation of mRNA into proteins, which include folding, post-translational modification, aggregation into secretory granules and transport outside the cell. One of the most important things to consider are the signal peptides, as they take a great role in directing the protein secretion. Unless the signal sequence is optimized, extracellular mRNA should be transfected ideally into cells that the encoded protein is naturally secreted in [174][44]. A table with current clinical trials on going is provided in this section (Table 3).
Table 3. mRNA Nanomedicines for Protein Replacement Therapies in Clinical Trials. A list of clinical trials for protein replacement applications is provided in this table.
| mRNA Delivery for Protein Replacement Therapies | |||||
|---|---|---|---|---|---|
| mRNA-3927 | Propionic Acidemia | Modified mRNA-LNPs | Phase 1/2 | NCT04159103 | Moderna |
| MRT5005 | Cystic Fibrosis | Modified mRNA-LNPs | Phase 1/2 | NCT03375047 | Translate Bio |
2.3.1. Cystic Fibrosis
Cystic fibrosis (CF) is a genetic disease which affects over 90,000 individuals all over the world. The F508del is the most prevalent CF-causing mutation, and affects approximately 82% of the CF population. This mutation leads to CFTR-protein misfolding, which is then arrested by the cell machinery and targeted to the proteasome, leading to its premature degradation [175][45]. Thus, mRNA-mediated replacement therapy can be applied to produce copies of the cystic fibrosis conductance receptor (CFTR), which is defective in CF patients. However, when it comes to delivery of mRNA nanoparticles to the lung, these nanoformulations not only need to be endocytosed, escape the endosome and release the cargo inside the cytoplasm, but they also need to migrate across viscous mucus, avoid clearance and uptake by the macrophages within this mucus and the airways and finally reach the pulmonary epithelial cells [175,176][45][46]. These additional biological barriers make the delivery of mRNA-loaded nanoparticles to the lung especially challenging. To date, the most advanced mRNA therapy for cystic fibrosis comes from Translate Bio and it is currently in Phase 1/2 (NCT03375047) [177][47].
2.3.2. Rare Metabolic Diseases
An important area which would be enormously impacted by this technology are rare metabolic diseases. These disorders lack financial support, and thus, effective treatments. From this point of view, mRNA technology can entail an enormous advantage due to the cost-efficient matters already commented. Furthermore, the previous idea and background given on endogenously produced proteins can also be applied in this section.
Acute Intermittent Porphyria
Acute intermittent porphyria is a chronic disease caused by the hepatic deficiency of porphobilinogen deaminase (PBGD), the enzyme which catalyzes the transformation of porphobilinogen into protoporphyrinogen during the heme synthesis pathway [178][48]. This causes the accumulation of the porphyrin precursors γ-aminolevulinic acid (ALA) and porphobilinogen (PBG), which entails acute neurovisceral attacks associated with a high production of potentially neurotoxic porphyrin precursors [178][48]. Treatments so far are composed of hemin replacement therapy, which restores the regulatory heme pool in the liver and suppresses the accumulation of precursors; however, these reductions occur days after the injection and the levels of enzymes should be monitored closely. Although this is an efficacious treatment, 5% of patients suffer recurrent attacks over the years. Prophylactic hemin treatment is being more commonly used for chronic symptoms; however, repeated administrations have been shown to have important side effects [179][49] RNA therapies seem to give an answer to the treatment of this disease. The siRNA drug Givosiran® aims to silence the ALAS1 enzyme, stopping the production of ALA and PBG. mRNA therapies are still in preclinical development. In this regard, Jiang et al. evaluated, along with the Moderna, the potential treatment of this disease with mRNA-encoding PBGD enzyme into LNPs in two models of acute hepatic porphyria. Furthermore, they also performed a multiple-dose study on non-human primates to further prove its translatability to humans. The results showed that administration of hPBGD mRNA led to expression of the therapeutic protein after 2 h post-administration, and most importantly, maintained its activity throughout the entire duration of the attack [180][50]. Overall, this sentudry reported sustained efficacy and tolerability of the hPBGD mRNA after single and repeated administrations in mice, rabbit and non-human primates [180][50], which further evidenced the potential of mRNA for the treatment of this disease.
Propionic Acidaemia (PA)
Propionic Acidaemia (PA) is a rare pediatric disorder caused by propionyl-CoA carboxylase (PCC) deficiency, which results in impaired propionate metabolism and accumulation of toxic metabolites, leading to a life-threatening condition. There are two disease subtypes, PCCA deficiency and PCCB deficiency, each of them referring to the different subunits forming PCC, respectively A (of 72 kDa) or B (of 54 kDa). Current treatments for this disease include fluid therapies or actions to remove the excess of toxic metabolites, but liver transplant is also a potential outcome for these patients. In this term, mRNA-therapeutic encoding for PCC enzymes can provide a solution. In this study, Jiang L. et al. report the use of two mRNAs encoding both human PCCA and PCCB. Results showed the restoration of ammonia levels and the function of the PCC enzyme in the liver; reducing toxic metabolites associated with this condition up to 6 months upon repeated administrations in mice [181][51]. Currently, mRNA-3927 from Moderna Therapeutics is under clinical trials (NCT04159103).
Fabry Disease
Fabry Disease is an X-linked lysosomal storage disorder which results from the mutation of the gene GLA encoding for the lysosomal enzyme α-galactosidase A, responsible for glycolipids’ metabolism. Its loss of activity leads to fat accumulation within the lysosomes of multiple tissues, which in turn leads to clinical disease with fatal renal and cardiac function alteration [182][52]. Current treatments for this metabolic disease include enzyme replacement therapies named Replagal® and Fabrazyme® [183][53]; and migalastat [184][54], a small molecule chaperone. While these treatments have shown to slow the progression of the disease, the variability of response, immunogenicity and difficulties with the manufacturing of these therapies are needs which have not yet been met. In this context, the main studies with mRNA have been published so far, showing some advancements. Using mRNA encoding for α-galactosidase, DeRosa et al. demonstrated it is possible to achieve sustained delivery in a Fabry model mouse and in a non-human primate with a resulting reduction of relevant biomarkers [185][55]. Upon delivery, serum protein levels reached as high as 1330-fold over normal physiological values. Another study conducted by Moderna also showed promising results upon intravenous administration of LNPs, with a dose-dependent protein activity and substrate reduction maintained for up to 6 weeks in mice. Upon repeated administration, the pharmacodynamic response was sustained and the administration to non-human primates confirmed safety and translatability [186][56].
Methylmalonic Acidemia (MMA)
Methylmalonic Acidemia (MMA) is a rare metabolic disease which is characterized by mutations in the gene coding of the vitamin B12-dependent enzyme methyl-malonyl-CoA (MUT), causing partial or complete loss of activity. This enzyme catalyzes important metabolic reactions linked to the Krebs cycle, playing a central role in intermediary metabolism. Its partial or total lack of activity causes the accumulation of toxic metabolites and the alteration of mitochondrial oxidative phosphorylation. Clinically, patients affected with this disorder experience multisystemic complications which are more common in organs with high metabolic demand, such as the brain or heart [187][57].
There is currently no effective treatment for these patients, and this disorder is often treated by carnitine supplementation and protein-limited diets; however, mortality is still around 20%, and survivors can develop systemic complications. mRNA therapy encoding for human MUT using LNPs as delivery vehicles was studied by An et al. in two clinically relevant models. In both, a decrease in the enzyme substrate methylmalonic acid blood levels were achieved, with good pharmacokinetic properties. Importantly, none of the clinical models developed any hMUT antibodies upon repeated administration or hepatic inflammation [188][58].
Ornithine Transcarbamylase Deficiency (OTD)
Ornithine transcarbamylase deficiency (OTD) affects 6 in 100,000 people, and is the most frequent inborn error of the urea cycle. As other urea-cycle disorders, it results in ammonia accumulation which clinically translates to fatal neurological disorders. OTD is more specifically caused by a deficiency in ornithine transcarbamylase (OTC). Therapeutic options for these patients include a protein-restricted diet for life, with arginine and citrulline supplementation, but liver transplantation is the only therapeutic option in cases of severe OTC deficiency [189][59]. In this study, the mRNA administration resulted in increased OTC enzymatic activity up to 10 days after dosing, along with increased mice survival [190][60].
2.4. Gene Editing
Clustered regularly interspaced short palindromic repeat (CRISPR)-related systems have become the main tool when considering therapeutic approaches for the cure of genetic diseases. CRISPR-Cas9 complex is composed of two main elements: Cas9 protein, an endonuclease that is capable of DNA cleaving; and a single-guide RNA (sgRNA), which guides the Cas9 protein to the site of interest. This gene-editing tool is, therefore, based on the recognition of PAM (protospacer-adjacent motif) sequence and a complementary 20-nucleotide genomic sequence that induces DNA brakes, which are repaired by error-prone nonhomologous end-joining or precise homology-directed repair [191][61]. To date, CRISPR/Cas9-based systems are versatile and have extensively been used for biotechnological purposes such as knockout/knock-in animal models or cells. From a drug-delivery point of view, it is important to understand that intracellular delivery of both components is needed. Therefore, various in vivo studies addressing differing delivery approaches such as viral vectors, mRNA or recombinant protein have been performed. In this section we will focus solely on mRNA delivery will be the focus, as it is the purpose of this reviewentry [191,192,193,194,195][61][62][63][64][65].
One of the approaches that have been most extensively researched are hepatic diseases. In 2016, a study on human hereditary tyrosinemia (HHT) showed slightly promising results on correcting the fumarylacetoacetate hydrolase (FAH) mutation. This was performed by the systemic delivery of Cas9 mRNA encapsulated in LNPs and sgRNA encapsulated in AAV particles. The final results demonstrated an overall gene correction of 6%, suggesting that this could be an effective approach for the treatment of a range of metabolic liver diseases. However, authors pointed out that one potential reason for the mild gene correction could be that the nanoformulations were not co-delivered, hypothesizing that the sgRNA suffered degradation during mRNA translation, and thus decreasing efficacy [195][65]. Following up on this, another study conducted by the same team aimed to deliver both Cas9 mRNA and sgRNA using LNPs for both components. In order to make it possible for the sgRNA to be efficiently delivered, they conducted several chemical modifications on the sequence, i.e., including the addition of 2’ hydroxyl (OH) groups on nucleotides, which resulted in highly improved results with an efficacy of 80% [196][66] This restudyearch is especially important as it highlights the importance of chemical modifications as well as the possibility of using synthetic nanoparticles, broadening up the industrial applications of gene editing. Similarly, Miller et al. reported successful results with a 95% protein knockdown in vitro, by making use of a zwitterionic lipid as a delivery vector by also co-delivering the different components [197][67] Adding to this latter study, Finn et al. reported a 97% reduction in serum protein levels upon the administration of Cas9 mRNA co-formulated with modified sgRNA into LNPs for gene editing of the mouse transthyretin gene in the liver [198][68]. To sum up, probably the most important advancement with regards to gene editing is the NTLA-2001 clinical study (NCT04601051), the first clinical trial so far with CRISPR-Cas9, conducted by Intellia Therapeutics for patients with hereditary ATTR amyloidosis suffering from polyneuropathy. This gene-editing medicine is based on the CRISPR Cas9 system and it uses mRNA-encoding Cas9 protein and the guide RNA encapsulated in a LNPs. Results showed mild adverse effects, and the administration led to a decrease in TTR after a single dose [199][69]. In October 2021, Intellia Therapeutics announced the beginning of a new CRISPR clinical trial, NTLA-2002, for the treatment of hereditary angioedema (HE), a rare autosomal dominant disorder that causes unexpected inflammatory attacks [200][70]. Altogether, these results present an undeniable precedent for gene editing. A current landscape on CRISPR-Cas9 clinical trials is provided next (Table 4).
Table 4. mRNA Nanomedicines for Gene editing in Clinical Trials. A list of clinical trials for gene editing applications is provided in this table. Abbreviations: sg, single-guide.
| Gene Editing | |||||
|---|---|---|---|---|---|
| NTLA-2001 | Hereditary Transthyretin Amyloidosis | Cas9 mRNA and sg mRNA-LNPs | Phase 1 | NCT04601051 | Intellia Therapeutics |
| NTLA-2002 | Hereditary Angioedema | Cas9 mRNA and sg mRNA LNPs | Phase 1/2 | Not registered yet | Intellia Therapeutics |
2.5. Autoimmune Diseases
Autoimmune diseases are chronic and debilitating conditions which are caused by immune response against self-antigens. Disorders such as multiple sclerosis or diabetes type I affect millions of people worldwide. Current treatments available do not target the origin of the disease and are not curative. However, immune tolerance—defined as the state of unresponsiveness of the immune system (IS) to any substance that has the potential to trigger a response—has been described before as a potential therapeutic tool [201,202][71][72]. In this term, mRNA technology could potentially be used to induce self-tolerance, by administering a self-antigen encoded in mRNA. In a recent study, Krienke et al. [203][73] showed that it is possible to deliver myelin oligodendrocyte glycoprotein (MOG)-encoded mRNA by making use of the lipoplex-type nanocarrier to multiple-sclerosis mice models. Upon intravenous administration, modified mRNA was capable of triggering the proliferation of T-reg cells without inducing immune activation, preventing myelin damage and paralysis [203][73]. This is, to the best of our knowledge, the first proof of concept using mRNA technology for immune tolerance.
2.6. Cardiovascular Diseases
Another potential for the development of mRNA therapeutics is treating myocardial infarction (MI) and heart failure (HF), the leading causes of death in many countries. MI leads to a massive loss of cardiomyocytes (CM), which are replaced by highly proliferating fibroblasts, resulting in loss of healthy tissue. These changes are irreversible, and along with oxidative stress and inflammation, the ultimate result is heart failure. Gene therapy with DNA-based or viral vectors has been used to try to induce cardiac regeneration, but has not had great success due to poor and uncontrolled gene expression. However, mRNA technology could give a plausible response for the treatment of ischemic injury, mainly by three different mechanisms: (1) inducing CM proliferation; (2) inhibiting heart cell death and attenuating inflammation; and (3) supporting cardiovascular regeneration. It is important to note that although mRNA applications in cardiology are progressing quickly, right now research is focused on intracardiac administration, as it is the most effective delivery method so far; however, it causes stress and local injury to the tissue. Therefore, it is crucial to develop a delivery vehicle that can target the heart and that can be then administered s.c. or i.v. [204,205][74][75]. mRNA encoding for vascular endothelial growth factor A (VEGF-A) is currently under clinical phase 2 (Table 5) in a collaboration between AstraZeneca and Moderna Therapeutics after showing the induction of cardiovascular regeneration in several studies. In addition, Magadum et al. were able to induce CM proliferation by using mRNA encoding for FSTL1 and Pkm2 [204][74].
Table 5. mRNA Nanomedicines for cardiovascular diseases in Clinical Trials. A list of clinical trials for cardiovascular diseases is provided in this table.
| Cardiovascular Diseases | |||||
|---|---|---|---|---|---|
| AZD8601 | Heart Failure | Modified mRNA-LNPs | Phase II | NCT03370887 | AstraZeneca |
References
- Gros, F.; Hiatt, H.; Gilbert, W.; Kurland, C.G.; Risebrough, R.W.; Watson, J.D. Unstable Ribonucleic Acid Revealed by Pulse Labelling of Escherichia Coli. Nature 1961, 190, 581–585.
- Brenner, S.; Jacob, F.; Meselson, M. An Unstable Intermediate Carrying Information from Genes to Ribosomes for Protein Synthesis. Nature 1961, 190, 576–581.
- Jacob, F.; Monod, J. Genetic Regulatory Mechanisms in the Synthesis of Proteins. J. Mol. Biol. 1961, 3, 318–356.
- Sahin, U.; Karikó, K.; Türeci, Ö. MRNA-Based Therapeutics—Developing a New Class of Drugs. Nat. Rev. Drug Discov. 2014, 13, 759–780.
- Weng, Y.; Li, C.; Yang, T.; Hu, B.; Zhang, M.; Guo, S.; Xiao, H.; Liang, X.-J.; Huang, Y. The Challenge and Prospect of MRNA Therapeutics Landscape. Biotechnol. Adv. 2020, 40, 107534.
- Hajj, K.A.; Whitehead, K.A. Tools for Translation: Non-Viral Materials for Therapeutic MRNA Delivery. Nat. Rev. Mater. 2017, 2, 17056.
- Pardi, N.; Hogan, M.J.; Porter, F.W.; Weissman, D. MRNA Vaccines—A New Era in Vaccinology. Nat. Rev. Drug Discov. 2018, 17, 261–279.
- Martinon, F.; Krishnan, S.; Lenzen, G.; Magné, R.; Gomard, E.; Guillet, J.-G.; Lévy, J.-P.; Meulien, P. Induction of Virus-Specific Cytotoxic T Lymphocytesin Vivo by Liposome-Entrapped MRNA. Eur. J. Immunol. 1993, 23, 1719–1722.
- Kowalski, P.S.; Rudra, A.; Miao, L.; Anderson, D.G. Delivering the Messenger: Advances in Technologies for Therapeutic MRNA Delivery. J. Soc. Gene Ther. 2019, 27, 710–728.
- Chaudhary, N.; Weissman, D.; Whitehead, K.A. MRNA Vaccines for Infectious Diseases: Principles, Delivery and Clinical Translation. Nat. Rev. Drug Discov. 2021, 20, 817–838.
- Dybul, M.; Attoye, T.; Baptiste, S.; Cherutich, P.; Dabis, F.; Deeks, S.G.; Dieffenbach, C.; Doehle, B.; Goodenow, M.M.; Jiang, A.; et al. The Case for an HIV Cure and How to Get There. Lancet HIV 2021, 8, e51–e58.
- Mascola, J.R. The Modern Era of HIV-1 Vaccine Development. Science 2015, 349, 139–140.
- Brito, L.A.; Chan, M.; Shaw, C.A.; Hekele, A.; Carsillo, T.; Schaefer, M.; Archer, J.; Seubert, A.; Otten, G.R.; Beard, C.W.; et al. A Cationic Nanoemulsion for the Delivery of Next-Generation RNA Vaccines. Mol. Ther. 2014, 22, 2118–2129.
- Bogers, W.M.; Oostermeijer, H.; Mooij, P.; Koopman, G.; Verschoor, E.J.; Davis, D.; Ulmer, J.B.; Brito, L.A.; Cu, Y.; Banerjee, K.; et al. Potent Immune Responses in Rhesus Macaques Induced by Nonviral Delivery of a Self-Amplifying RNA Vaccine Expressing HIV Type 1 Envelope with a Cationic Nanoemulsion. J. Infect. Dis. 2015, 211, 947–955.
- Pollard, C.; Rejman, J.; de Haes, W.; Verrier, B.; van Gulck, E.; Naessens, T.; de Smedt, S.; Bogaert, P.; Grooten, J.; Vanham, G.; et al. Type I IFN Counteracts the Induction of Antigen-Specific Immune Responses by Lipid-Based Delivery of MRNA Vaccines. Mol. Ther. 2013, 21, 251–259.
- Zhang, P.; Narayanan, E.; Liu, Q.; Tsybovsky, Y.; Boswell, K.; Ding, S.; Hu, Z.; Follmann, D.; Lin, Y.; Miao, H.; et al. A Multiclade Env–Gag VLP MRNA Vaccine Elicits Tier-2 HIV-1-Neutralizing Antibodies and Reduces the Risk of Heterologous SHIV Infection in Macaques. Nat. Med. 2021, 27, 2234–2245.
- Pardi, N.; Secreto, A.J.; Shan, X.; Debonera, F.; Glover, J.; Yi, Y.; Muramatsu, H.; Ni, H.; Mui, B.L.; Tam, Y.K.; et al. Administration of Nucleoside-Modified MRNA Encoding Broadly Neutralizing Antibody Protects Humanized Mice from HIV-1 Challenge. Nat. Commun. 2017, 8, 14630.
- Kaner, J.; Schaack, S. Understanding Ebola: The 2014 Epidemic. Glob. Health 2016, 12, 1–7.
- Meyer, M.; Huang, E.; Yuzhakov, O.; Ramanathan, P.; Ciaramella, G.; Bukreyev, A. Modified MRNA-Based Vaccines Elicit Robust Immune Responses and Protect Guinea Pigs From Ebola Virus Disease. J. Infect. Dis. 2018, 217, 451–455.
- WHO. WHO Recommends Groundbreaking Malaria Vaccine for Children at Risk; WHO: Geneva, Switzerland, 2021.
- Baeza Garcia, A.; Siu, E.; Sun, T.; Exler, V.; Brito, L.; Hekele, A.; Otten, G.; Augustijn, K.; Janse, C.J.; Ulmer, J.B.; et al. Neutralization of the Plasmodium-Encoded MIF Ortholog Confers Protective Immunity against Malaria Infection. Nat. Commun. 2018, 9, 2714.
- Sun, T.; Holowka, T.; Song, Y.; Zierow, S.; Leng, L.; Chen, Y.; Xiong, H.; Griffith, J.; Nouraie, M.; Thuma, P.E.; et al. A Plasmodium-Encoded Cytokine Suppresses T-Cell Immunity during Malaria. Proc. Natl. Acad. Sci. USA 2012, 109, E2117–E2126.
- Raj, D.K.; das Mohapatra, A.; Jnawali, A.; Zuromski, J.; Jha, A.; Cham-Kpu, G.; Sherman, B.; Rudlaff, R.M.; Nixon, C.E.; Hilton, N.; et al. Anti-PfGARP Activates Programmed Cell Death of Parasites and Reduces Severe Malaria. Nature 2020, 582, 104–108.
- Goodall, G.J.; Wickramasinghe, V.O. RNA in Cancer. Nat. Rev. Cancer 2021, 21, 22–36.
- Pan, C.; Liu, H.; Robins, E.; Song, W.; Liu, D.; Li, Z.; Zheng, L. Next-Generation Immuno-Oncology Agents: Current Momentum Shifts in Cancer Immunotherapy. J. Hematol. Oncol. 2020, 13, 29.
- Upadhaya, S.; Hubbard-Lucey, V.M.; Yu, J.X. Immuno-Oncology Drug Development Forges on despite COVID-19. Nat. Rev. Drug Discov. 2020, 19, 751–752.
- Van Lint, S.; Renmans, D.; Broos, K.; Goethals, L.; Maenhout, S.; Benteyn, D.; Goyvaerts, C.; Four, S.; van der Jeught, K.; Bialkowski, L.; et al. Intratumoral Delivery of TriMix MRNA Results in T-Cell Activation by Cross-Presenting Dendritic Cells. Cancer Immunol. Res. 2016, 4, 146–156.
- Irvine, D.J.; Dane, E.L. Enhancing Cancer Immunotherapy with Nanomedicine. Nat. Rev. Immunol. 2020, 20, 321–334.
- Pastor, F.; Berraondo, P.; Etxeberria, I.; Frederick, J.; Sahin, U.; Gilboa, E.; Melero, I. An RNA Toolbox for Cancer Immunotherapy. Nat. Rev. Drug Discov. 2018, 17, 751–767.
- Blass, E.; Ott, P.A. Advances in the Development of Personalized Neoantigen-Based Therapeutic Cancer Vaccines. Nat. Rev. Clin. Oncol. 2021, 18, 215–229.
- Miao, L.; Zhang, Y.; Huang, L. MRNA Vaccine for Cancer Immunotherapy. Mol. Cancer 2021, 20, 1–23.
- Rosenblum, D.; Joshi, N.; Tao, W.; Karp, J.M.; Peer, D. Progress and Challenges towards Targeted Delivery of Cancer Therapeutics. Nat. Commun. 2018, 9, 1410.
- van Hoecke, L.; Roose, K. How MRNA Therapeutics Are Entering the Monoclonal Antibody Field. J. Transl. Med. 2019, 17, 54.
- Hewitt, S.L.; Bai, A.; Bailey, D.; Ichikawa, K.; Zielinski, J.; Karp, R.; Apte, A.; Arnold, K.; Zacharek, S.J.; Iliou, M.S.; et al. Durable Anticancer Immunity from Intratumoral Administration of IL-23, IL-36γ, and OX40L MRNAs. Sci. Transl. Med. 2019, 11, eaat9143.
- Berraondo, P.; Etxeberria, I.; Ponz-Sarvise, M.; Melero, I. Revisiting Interleukin-12 as a Cancer Immunotherapy Agent. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2018, 24, 2716–2718.
- Lai, I.; Swaminathan, S.; Baylot, V.; Mosley, A.; Dhanasekaran, R.; Gabay, M.; Felsher, D.W. Lipid Nanoparticles That Deliver IL-12 Messenger RNA Suppress Tumorigenesis in MYC Oncogene-Driven Hepatocellular Carcinoma. J. Immunother. Cancer 2018, 6, 125.
- Li, Y.; Su, Z.; Zhao, W.; Zhang, X.; Momin, N.; Zhang, C.; Wittrup, K.D.; Dong, Y.; Irvine, D.J.; Weiss, R. Multifunctional Oncolytic Nanoparticles Deliver Self-Replicating IL-12 RNA to Eliminate Established Tumors and Prime Systemic Immunity. Nat. Cancer 2020, 1, 882–893.
- Berraondo, P.; Sanmamed, M.F.; Ochoa, M.C.; Etxeberria, I.; Aznar, M.A.; Pérez-Gracia, J.L.; Rodríguez-Ruiz, M.E.; Ponz-Sarvise, M.; Castañón, E.; Melero, I. Cytokines in Clinical Cancer Immunotherapy. Br. J. Cancer 2019, 120, 6–15.
- Rybakova, Y.; Kowalski, P.S.; Huang, Y.; Gonzalez, J.T.; Heartlein, M.W.; DeRosa, F.; Delcassian, D.; Anderson, D.G. MRNA Delivery for Therapeutic Anti-HER2 Antibody Expression In Vivo. Mol. Ther. 2019, 27, 1415–1423.
- Stadler, C.R.; Bähr-Mahmud, H.; Celik, L.; Hebich, B.; Roth, A.S.; Roth, R.P.; Karikó, K.; Türeci, Ö.; Sahin, U. Elimination of Large Tumors in Mice by MRNA-Encoded Bispecific Antibodies. Nat. Med. 2017, 23, 815–817.
- Han, X.; Wang, Y.; Han, W.-D. Chimeric Antigen Receptor Modified T-Cells for Cancer Treatment. Chronic Dis. Transl. Med. 2018, 4, 225–243.
- Scarfò, I.; Maus, M.V. Current Approaches to Increase CAR T Cell Potency in Solid Tumors: Targeting the Tumor Microenvironment. J. ImmunoTherapy Cancer 2017, 5, 28.
- Reinhard, K.; Rengstl, B.; Oehm, P.; Michel, K.; Billmeier, A.; Hayduk, N.; Klein, O.; Kuna, K.; Ouchan, Y.; Wöll, S.; et al. An RNA Vaccine Drives Expansion and Efficacy of Claudin-CAR-T Cells against Solid Tumors. Science 2020, 367, 446.
- Berraondo, P.; Martini, P.G.; Avila, M.A.; Fontanellas, A. Messenger RNA Therapy for Rare Genetic Metabolic Diseases. Gut 2019, 68, 1323.
- Jaques, R.; Shakeel, A.; Hoyle, C. Novel Therapeutic Approaches for the Management of Cystic Fibrosis. Multidiscip. Respir. Med. 2020, 15, 690.
- Christopher Boyd, A.; Guo, S.; Huang, L.; Kerem, B.; Oren, Y.S.; Walker, A.J.; Hart, S.L. New Approaches to Genetic Therapies for Cystic Fibrosis. J. Cyst. Fibros. 2020, 19, S54–S59.
- Sanchez, A.D.S.; Paunovska, K.; Cristian, A.; Dahlman, J.E. Treating Cystic Fibrosis with MRNA and CRISPR. Hum. Gene Ther. 2020, 31, 940–955.
- Bissell, D.M.; Anderson, K.E.; Bonkovsky, H.L. Porphyria. N. Engl. J. Med. 2017, 377, 862–872.
- Anderson, K.E.; Bloomer, J.R.; Bonkovsky, H.L.; Kushner, J.P.; Pierach, C.A.; Pimstone, N.R.; Desnick, R.J. Recommendations for the Diagnosis and Treatment of the Acute Porphyrias. Ann Intern Med. 2005, 142, 439–450.
- Jiang, L.; Berraondo, P.; Jericó, D.; Guey, L.T.; Sampedro, A.; Frassetto, A.; Benenato, K.E.; Burke, K.; Santamaría, E.; Alegre, M.; et al. Systemic Messenger RNA as an Etiological Treatment for Acute Intermittent Porphyria. Nat. Med. 2018, 24, 1899–1909.
- Jiang, L.; Park, J.-S.; Yin, L.; Laureano, R.; Jacquinet, E.; Yang, J.; Liang, S.; Frassetto, A.; Zhuo, J.; Yan, X.; et al. Dual MRNA Therapy Restores Metabolic Function in Long-Term Studies in Mice with Propionic Acidemia. Nat. Commun. 2020, 11, 5339.
- Chan, B.; Adam, D.N. A Review of Fabry Disease. Ski. Ther. Lett. 2018, 23, 4–6.
- Pastores, G.M. Agalsidase Alfa (Replagal) in the Treatment of Anderson-Fabry Disease. Biol. Targets Ther. 2007, 1, 291–300.
- Germain, D.P.; Hughes, D.A.; Nicholls, K.; Bichet, D.G.; Giugliani, R.; Wilcox, W.R.; Feliciani, C.; Shankar, S.P.; Ezgu, F.; Amartino, H.; et al. Treatment of Fabry’s Disease with the Pharmacologic Chaperone Migalastat. N. Engl. J. Med. 2016, 375, 545–555.
- DeRosa, F.; Smith, L.; Shen, Y.; Huang, Y.; Pan, J.; Xie, H.; Yahalom, B.; Heartlein, M.W. Improved Efficacy in a Fabry Disease Model Using a Systemic MRNA Liver Depot System as Compared to Enzyme Replacement Therapy. Mol. Ther. J. Am. Soc. Gene Ther. 2019, 27, 878–889.
- Zhu, X.; Yin, L.; Theisen, M.; Zhuo, J.; Siddiqui, S.; Levy, B.; Presnyak, V.; Frassetto, A.; Milton, J.; Salerno, T.; et al. Systemic MRNA Therapy for the Treatment of Fabry Disease: Preclinical Studies in Wild-Type Mice, Fabry Mouse Model, and Wild-Type Non-Human Primates. Am. J. Hum. Genet. 2019, 104, 625–637.
- Fraser, J.L.; Venditti, C.P. Methylmalonic and Propionic Acidemias: Clinical Management Update. Curr. Opin. Pediatrics 2016, 28, 682–693.
- An, D.; Schneller, J.L.; Frassetto, A.; Liang, S.; Zhu, X.; Park, J.-S.; Theisen, M.; Hong, S.-J.; Zhou, J.; Rajendran, R.; et al. Systemic Messenger RNA Therapy as a Treatment for Methylmalonic Acidemia. Cell Rep. 2017, 21, 3548–3558.
- Peng, M.-Z.; Li, X.-Z.; Mei, H.-F.; Sheng, H.-Y.; Yin, X.; Jiang, M.-Y.; Cai, Y.-N.; Su, L.; Lin, Y.-T.; Shao, Y.-X.; et al. Clinical and Biochemical Characteristics of Patients with Ornithine Transcarbamylase Deficiency. Clin. Biochem. 2020, 84, 63–72.
- Prieve, M.G.; Harvie, P.; Monahan, S.D.; Roy, D.; Li, A.G.; Blevins, T.L.; Paschal, A.E.; Waldheim, M.; Bell, E.C.; Galperin, A.; et al. Targeted MRNA Therapy for Ornithine Transcarbamylase Deficiency. J. Am. Soc. Gene Ther. 2018, 26, 801–813.
- Doudna, J.A.; Charpentier, E. Genome Editing. The New Frontier of Genome Engineering with CRISPR-Cas9. Science 2014, 346, 1258096.
- Luther, D.C.; Lee, Y.W.; Nagaraj, H.; Scaletti, F.; Rotello, V.M. Delivery Approaches for CRISPR/Cas9 Therapeutics in Vivo: Advances and Challenges. null 2018, 15, 905–913.
- Mali, P.; Esvelt, K.M.; Church, G.M. Cas9 as a Versatile Tool for Engineering Biology. Nat. Methods 2013, 10, 957–963.
- Sander, J.D.; Joung, J.K. CRISPR-Cas Systems for Editing, Regulating and Targeting Genomes. Nat. Biotechnol. 2014, 32, 347–355.
- Yin, H.; Song, C.-Q.; Dorkin, J.R.; Zhu, L.J.; Li, Y.; Wu, Q.; Park, A.; Yang, J.; Suresh, S.; Bizhanova, A.; et al. Therapeutic Genome Editing by Combined Viral and Non-Viral Delivery of CRISPR System Components in Vivo. Nat. Biotechnol. 2016, 34, 328–333.
- Yin, H.; Song, C.-Q.; Suresh, S.; Wu, Q.; Walsh, S.; Rhym, L.H.; Mintzer, E.; Bolukbasi, M.F.; Zhu, L.J.; Kauffman, K.; et al. Structure-Guided Chemical Modification of Guide RNA Enables Potent Non-Viral in Vivo Genome Editing. Nat. Biotechnol. 2017, 35, 1179–1187.
- Miller, J.B.; Zhang, S.; Kos, P.; Xiong, H.; Zhou, K.; Perelman, S.S.; Zhu, H.; Siegwart, D.J. Non-Viral CRISPR/Cas Gene Editing In Vitro and In Vivo Enabled by Synthetic Nanoparticle Co-Delivery of Cas9 MRNA and SgRNA. Angew. Chem. 2017, 56, 1059–1063.
- Finn, J.D.; Smith, A.R.; Patel, M.C.; Shaw, L.; Youniss, M.R.; van Heteren, J.; Dirstine, T.; Ciullo, C.; Lescarbeau, R.; Seitzer, J.; et al. A Single Administration of CRISPR/Cas9 Lipid Nanoparticles Achieves Robust and Persistent In Vivo Genome Editing. Cell Rep. 2018, 22, 2227–2235.
- Gillmore, J.D.; Gane, E.; Taubel, J.; Kao, J.; Fontana, M.; Maitland, M.L.; Seitzer, J.; O’Connell, D.; Walsh, K.R.; Wood, K.; et al. CRISPR-Cas9 In Vivo Gene Editing for Transthyretin Amyloidosis. N. Engl. J. Med. 2021, 385, 493–502.
- Intellia Therapeutics Intellia Therapeutics. Available online: https://ir.intelliatx.com/news-releases/news-release-details/intellia-therapeutics-receives-authorization-initiate-phase-12 (accessed on 16 October 2021).
- Rayner, F.; Isaacs, J.D. Therapeutic Tolerance in Autoimmune Disease. Semin. Arthritis Rheum. 2018, 48, 558–562.
- Serra, P.; Santamaria, P. Antigen-Specific Therapeutic Approaches for Autoimmunity. Nat. Biotechnol. 2019, 37, 238–251.
- Krienke, C.; Kolb, L.; Diken, E.; Streuber, M.; Kirchhoff, S.; Bukur, T.; Akilli-Öztürk, Ö.; Kranz, L.M.; Berger, H.; Petschenka, J.; et al. A Noninflammatory MRNA Vaccine for Treatment of Experimental Autoimmune Encephalomyelitis. Science 2021, 371, 145–153.
- Magadum, A.; Kaur, K.; Zangi, L. MRNA-Based Protein Replacement Therapy for the Heart. Mol. Ther. 2019, 27, 785–793.
- Kaur, K.; Zangi, L. Modified MRNA as a Therapeutic Tool for the Heart. Cardiovasc. Drugs Ther. 2020, 34, 871–880.
More