Cardiovascular diseases (CVDs) are still a major cause of global mortality and disability, seriously affecting people’s lives. Due to the severity and complexity of these diseases, it is important to find new regulatory mechanisms to treat CVDs. Ferroptosis is a new kind of regulatory cell death currently being investigated. Increasing evidence showed that ferroptosis plays an important role in CVDs, such as in ischemia/reperfusion injury, heart failure, cardiomyopathy, and atherosclerosis. 心血管疾病(CVDs)仍然是全球死亡和残疾的主要原因,严重影响人们的生活。由于这些疾病的严重性和复杂性,寻找新的调节机制来治疗心血管疾病非常重要。铁死亡是目前正在研究的一种新型调节性细胞死亡。越来越多的证据表明,铁死亡在心血管疾病中起着重要作用,例如在缺血/再灌注损伤、心力衰竭、心肌病和动脉粥样硬化中。通过靶向铁死亡来预防心血管疾病是一种很有前途的方法;因此,在这篇综述中,我们总结了铁死亡的最新调控机制和目前与每种 CVD 相关的研究,然后对 CVD 的铁死亡治疗以及这一有趣生物学的未来方向提出了批判性观点。
1. Introduction一、简介
Ferroptosis 是一种新型的细胞死亡,起源于拉丁语“ferrum”和希腊语“ptosis”,分别意为“铁”和“坠落”[ 1 ]。Dixon 和他的同事在 2012 年首次报道它是一种铁依赖性的非凋亡性细胞死亡形式 [ 2 ]。它的特点是致密、紧凑的线粒体,嵴缺失,这是独特的,与其他形式的细胞死亡不同,如细胞凋亡(染色质边缘和浓缩)、坏死(破裂的质膜和肿胀的细胞质)和自噬(封闭双膜囊泡形成) [ 3 , 4]。铁死亡被确定参与肿瘤、神经退行性疾病(阿尔茨海默病、帕金森病、亨廷顿病等)、脑卒中、缺血/再灌注(I/R)等多种疾病,成为研究热点和重点,值得进一步探索。
Ferroptosis, a new kind of cell death, originates from the Latin word “ferrum” and the Greek word “ptosis”, meaning “iron” and “a fall”, respectively [1]. Dixon and his colleagues first reported it in 2012 as an iron-dependent form of nonapoptotic cell death [2]. It is characterized by dense, compact mitochondria with a loss of crista, which is unique and distinct from other forms of cell deaths, such as apoptosis (chromatin margination and condensation), necrosis (ruptured plasma membrane and swollen cytoplasm), and autophagy (enclosed double-membrane vesicles formation) [3][4]. Ferroptosis was determined to participate in many diseases such as tumors, neurodegenerative disorders (Alzheimer’s disease, Parkinson’s disease, Huntington’s disease etc.), stroke, and ischemia/reperfusion (I/R), and it became a hotspot and focus of research, meriting further exploration.
Cardiovascular diseases (CVDs), a major cause of global mortality and disability, greatly affect people’s lives [5]. One ongoing multinational collaboration study shows that the burden of CVDs was increasing in nearly all countries [6]. The age-standardized rate of CVDs is even increasing in some high-income countries that previously had a declining rate [7]. Recent research demonstrated that ferroptosis participates in several CVDs, such as I/R [8][9][10][11], heart failure (HF) [12][13], cardiomyopathy [14][15], and atherosclerosis [16]. Given the huge potential of ferroptosis in the treatment of CVDs, the latest work should be summarized, together with a report on the progress of possible medical usage.
心血管疾病 (CVDs) 是全球死亡和残疾的主要原因,极大地影响着人们的生活 [ 5 ]。一项正在进行的跨国合作研究表明,几乎所有国家的 CVD 负担都在增加 [ 6 ]。在一些高收入国家,心血管疾病的年龄标准化率甚至在上升,而这些国家的发病率以前有所下降 [ 7 ]。最近的研究表明,铁死亡参与多种心血管疾病,例如 I/R [ 8 , 9 , 10 , 11 ]、心力衰竭 (HF) [ 12 , 13 ]、心肌病 [ 14 , 15 ] 和动脉粥样硬化 [ 16 ]]。鉴于铁死亡在 CVD 治疗中的巨大潜力,应总结最新工作,并报告可能的医疗用途进展情况。
2. The Regulating Pathways of Ferroptosis2. 铁死亡的调节途径
Ferroptosis was first proposed by Dixon as an iron-dependent regulated cell death
[17]. Triggered by small specific molecules such as erastin, a small RAS gene selective molecule, and RAS-selective lethal 3 (RSL3), ferroptosis is characterized by the unique morphological changes of mitochondria. The overproduction of lipid peroxides, not clearing in time, increases membrane permeability, reduces fluidity, and destroys ionic homeostasis; thus, affecting the normal structure and function of membrane, ultimately leading to cell death
[18]. Iron metabolism is critical in the formation of lipid peroxides
[19]. Xc-GSH-GPX4 pathway
[20], FSP1–CoQ10–NAD(P)H pathway
[21][22][21,22], GCH1–BH4–DHFR pathway
[23][24][23,24], and mitochondria
[25] all play an important role in scavenging excess lipid peroxides and regulating the process of ferroptosis (
Figure 1).
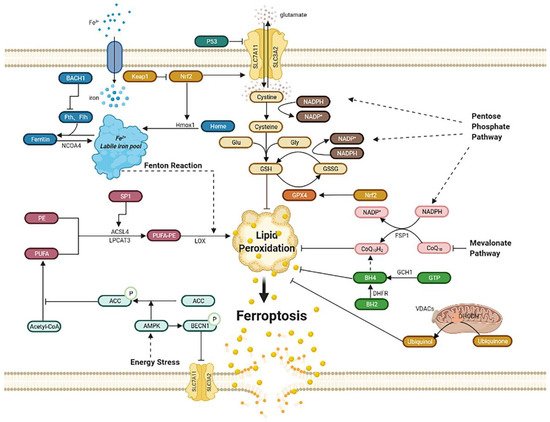
Figure 1. Regulatory pathways of ferroptosis. Ferroptosis is an iron-dependent, new kind of cell death. Regulatory pathways are complex, which can mainly be divided into several parts: iron metabolism; lipid peroxidation; Xc-GSH-GPX4 pathway; FSP1–CoQ10–NAD(P)H pathway; GCH1–BH4–DHFR pathway. Mitochondria and energy stress also play an important role. Abbreviations: BACH1: BTB domain and CNC homolog 1; Fth1: ferritin heavy chain 1; Ftl1: ferritin light chain 1; NCOA4: nuclear receptor coactivator 4; Hmox1: heme oxygenase-1; Keap1: Kelch-like ECH associated protein 1; Nrf2: nuclear factor erythroid 2-related factor 2; PE: phosphatidyl ethanolamine; PUFA: poly-unsaturated fatty acid; PUFA-PE: poly-unsaturated fatty acid-phosphatidyl ethanolamine; Sp1: special protein 1; ACSL4: acyl-CoA synthetase long-chain family member 4; LPCAT3: lysophosphatidylcholine acyltransferase 3; LOX: lipoxygenase; ACC: acetyl-CoA carboxylase; AMPK: AMP-activated protein kinase; BECN1: beclin 1; SLC7A11: subunit solute carrier family 7 member 11; SLC3A2: solute carrier family 3 member 2; Glu: glutamate; Gly: glycine; GSH: glutathione; GSSG: oxidized glutathione; GPX4: glutathione peroxidase 4; GCH1: guanosine triphosphate cyclohydrolase 1; GTP: guanosine triphosphate; BH4: tetrahydrobiopterin; BH2: dihydrobiopterin; DHFR: dihydrofolate reductase; DHODH: dihydroorotate dehydrogenase; VDACs: voltage-dependent anion channels; FSP1: ferroptosis suppressor protein1.
2.1. Iron Metabolism
Iron overload can lead to ferroptosis. Iron, the most abundant trace element in vivo, is regulated to maintain appropriate levels. Apart from being part of the composition of the human body, iron can be detrimental due to its active redox capacity, such as Fenton and Fenton-like reactions, which means ferrous iron (Fe2+) can react with oxygen or hydrogen peroxide (H
2O
2) and produce plenty of hydroxyl radicals and lipid peroxides, thus ultimately leading to ferroptosis
[19]; therefore, regulating iron homeostasis is of great importance. Iron homeostasis is related to nuclear receptor coactivator 4 (NCOA4)-mediated ferritinophagy, a kind of autophagy that turns ferritin into intracellular iron
[26][27][28][29][26,27,28,29]. Heme oxygenase-1 (Hmox1) also contributes to iron homeostasis, which exerts the degradation of heme and produces ferrous iron
[30][31][30,31]. The activation of ferritinophagy or overexpression of Hmox1can increases the free iron level, leading to lipid peroxides accumulation and ultimately ferroptosis. In addition, BTB domain and CNC homolog 1 (BACH1), a transcription factor, can promote ferroptosis by downregulating ferritin genes, including ferritin heavy chain 1 (Fth1) and ferritin light chain 1 (Ftl1), which reduce the amount of free labile iron. Therefore, it is possible to inhibit BACH1 to reduce free labile iron and further alleviate ferroptosis
[32].
2.2. Lipid Peroxidation
Ferroptosis can be induced by lipid peroxidation, consisting of nonenzymatic lipid peroxidation like Fenton reaction and enzymatic lipid peroxidation mediated by the activity of the lipoxygenase (LOX) family. Poly-unsaturated fatty acid-phosphatidyl ethanolamine (PUFA-PE) can be metabolized by Fe2+ and LOX to produce lipid peroxides. Poly-unsaturated fatty acid (PUFA), especially arachidonic acid (AA) and adrenaline, can be catalyzed by acyl-CoA synthetase long-chain family member 4 (ACSL4) and lysophosphatidylcholine acyltransferase 3 (LPCAT3), and they then take part in the biosynthesis of PUFA-PE with phosphatidylethanolamine (PE). Once the generation of lipid peroxides is hyperactive, lasting depletion of PUFA affects the normal structure and function of membrane and eventually leads to cell death
[19]. ACSL4 is identified as a pivotal biomarker of ferroptosis. Knocking down ACSL4 by shRNA inhibits ferroptosis, whereas the overexpression of ASCL4 contributes to the sensitivity of ferroptosis by modulating the cellular lipid composition
[33]. Special protein 1 (Sp1), an important factor, activates ACSL4 transcription when binding to the promoter region of ACSL4
[34]; consequently, increasing the expression or catalytic activity of ASCL4, LPCAT3, LOX, or Fenton reaction leads to lipid peroxides accumulation and, ultimately, ferroptosis
[35].
2.3. Xc-GSH-GPX4 Pathway
The Xc-GSH-GPX4 pathway is the main pathway to regulate ferroptosis. Ferroptosis is mainly induced by a large number of reactive oxygen species (ROS), far beyond the capability of clearing mechanisms. The main clearing mechanism that reduces ROS is the redox ability of glutathione (GSH). GSH, a tripeptide that includes glutamic acid, cysteine, and glycine, acts as an antioxidant and is the substrate of glutathione peroxidase 4 (GPX4), which is then converted into oxidized glutathione (GSSG). Increasing the expression of GSH can inhibit ferroptosis. Furthermore, the System Xc−, composed of subunit solute carrier family 7 member 11 (SLC7A11) and solute carrier family 3 member 2 (SLC3A2), mediates the exchange of cystine and glutamate across the plasma membrane at the ratio of 1:1 to synthesize GSH. The inhibition of System Xc− by erastin can lead to ferroptosis due to GSH depletion. This inhibition may cause the complete opposite effect of inhibiting ferroptosis. Inhibiting SLC7A11 decreases the amount of cystine and causes NADPH “debt” to reduce cystine to cysteine, thus impairing antioxidant ability and driving cells to ferroptosis
[36]. As for GPX4, it is a kind of selenoenzyme that reduces ROS through GSH
[20]. The inhibition of GPX4 by RSL3 leads to an impairment of antioxidant capacity, and then to ferroptosis.
Moreover, P53 can also regulate ferroptosis in two diametrically opposed ways
[37]. On the one hand, some studies report that P53 can lower the expression of SLC7A11, which affects the cystine transport function of system Xc- and further inhibits the activity of GPX4, ultimately leading to ferroptosis
[38]. On the other hand, P53 is reported to inhibit ferroptosis in some cells, involving cyclin-dependent kinase inhibitor 1 A (CDKN1A), a p53 transcriptional target
[39].
Furthermore, nuclear factor erythroid 2-related factor 2 (Nrf2) increases the level of SLC7A11 and transcriptionally induces the expression of GPX4, which then reduces ROS; therefore, the overexpression of Nrf2 can inhibit ferroptosis, whereas the Kelch-like ECH associated protein 1 (Keap1), bonding to Nrf2 and negatively regulating it, can reverse this process and exert a proferroptosis role
[40][41][40,41]. However, Nrf2 can also induce ferroptosis by upregulating Hmox1 then degrading heme and releasing free iron, which ultimately leads to ferroptosis
[14].
Cystine starvation can lead to ferroptosis because cysteine is crucial to GSH synthesis as a rate-limiting substrate, and the followed glutamate accumulation increases ROS. Interestingly, in nonsmall-cell lung cancer (NSCLC) cells, cystine starvation inducing ferroptosis can be inhibited by the generation of γ-glutamyl-peptide for reducing glutamate stress, which is catalyzed by glutamate–cysteine ligase catalytic subunit (GCLC), a Nrf2 regulating protein
[42]. In high-fat diet-induced nonalcoholic fatty liver disease (NAFLD), ferroptosis contributes its development, and ginkgolide B (GB) is effective for treatment by inhibiting lipid accumulation and oxidative stress, which are probably related to Nrf2 elevation induced by GB
[43]. All these show that Nrf2 is a crucial target regarding ferroptosis.
2.4. FSP1–CoQ10–NAD(P)H Pathway
Ferroptosis suppressor protein 1 (FSP1), previously named apoptosis-inducing factor mitochondria-associated 2 (AIFM2), is thought to inhibit ferroptosis as an independent system that co-operates with the Xc-GSH-GPX4 pathway
[21][22][21,22]. That means FSP1 is capable to eliminate lethal lipid peroxides even in the absence of GPX4. Acting as an oxidoreductase, FSP1 reduces ubiquinone (also named coenzyme Q10) into ubiquinol by NADPH, another antioxidant compound besides GSH, which can inhibit ferroptosis by lessening lipid ROS. Moreover, inhibiting the mevalonate pathway may decrease the amount of coenzyme Q10, thus leading to ferroptosis
[44] However, a study also showed that ubiquinol production is not essential for ferroptosis resistance mediated by FSP1, while endosomal sorting complexes required for transport III(ESCRT-III)–dependent membrane repair is responsible for it
[45]. Anyway, they all elaborate that FSP1 is a potential target for ferroptosis-related diseases.
2.5. Mitochondria
The dramatic morphological features of ferroptosis are the dense, compact mitochondria with the loss of crista. It is unique and distinct from other forms of cell death, such as apoptosis (chromatin margination and condensation), necrosis (ruptured plasma membrane and swollen cytoplasmic), and autophagy (enclosed double-membrane vesicles formation)
[46]. Voltage-dependent anion channels (VDACs), the ample protein located in the outer mitochondrial membrane, are essential to ferroptosis as a potential erastin target. The more VDACs proteins cells exist, the more sensitivity these cells exhibit to ferroptosis
[47]. Moreover, the mechanism of mitochondria participating in ferroptosis involves cystine starvation and glutaminolysis, which raise the content of membrane potential (MMP) and ferrous iron content and contribute to Fenton reaction and lipid peroxidation, leading to ferroptosis
[48][49][48,49].
Dihydroorotate dehydrogenase (DHODH), found on the inner membrane of mitochondrial, is reported to generate ubiquinol from ubiquinone by modulating the transformation of dihydroorotate to orotate, which demonstrates as a parallel way from mitochondrial GPX4 to protect cells from lipid peroxidation. Moreover, GPX4 knocks down sensitizing human tumor cells to DHODH inhibition. DHODH inhibitor maybe the potent anticancer agent in low GPX4 expression cancer. This mechanism of DHODH in mitochondrial is similar to the FSP1 system in cytosolic. Overexpression of FSP1 on mitochondria cannot affect lipid peroxidation, suggesting that the ferroptosis regulating the function of FSP1 may require some proteins on the plasma membrane
[25].
2.6. GCH1–BH4–DHFR Pathway
The guanosine triphosphate cyclohydrolase 1 (GCH1)–tetrahydrobiopterin (BH4)–dihydrofolate reductase (DHFR) pathway is another regulatory mechanism independently from Xc-GSH-GPX4 axis. Under the induction of ferroptosis by RSL3, erastin, and Gpx4 knockout, GCH1 exerts strong protective effect, which is selective from other forms of cell death
[23]. The upregulation of GCH1 increases the content of BH4, an endogenous antioxidant, which captures lipid-derived peroxyl radicals to block the process of lipid peroxidation. Moreover, DHFR is the most efficient way for the regeneration of BH4
[24]; therefore, GCH1–BH4–DHFR pathway inhibits ferroptosis by subverting lipid peroxidation.
2.7. Energy Stress
Energy stress is one of metabolic stress, characterized by the increase in intracellular AMP and the consumption of intracellular ATP. Adenosine monophosphate (AMP)-activated protein kinase (AMPK) is crucial to energy stress by stimulating the ATP-producing catabolic processes and inhibits ATP-consuming anabolic processes
[50]. Activation of AMPK in energy stress increases phosphorylation of acetyl-CoA carboxylase (ACC), thus hindering the synthesis and oxidation of fatty acids, which illustrates how energy stress inhibits ferroptosis
[51]. A study also reports that AMPK promotes ferroptosis by phosphorylating BECN1 (beclin 1), binding to SLC7A11, then inhibiting cystine transport
[52]; therefore, the precise effect of energy stress or AMPK needs further investigation.
3. Ferroptosis in Cardiovascular Diseases
3.1. Ferroptosis in Ischemia/Reperfusion (I/R)
Inhibiting ferroptosis can reduce I/R injury. I/R and ferroptosis were studied in several organs, such as the heart
[8][9][10][11][29][8,9,10,11,29], kidney
[51][53][54][55][51,66,67,68], brain
[56][69], intestine
[8], and liver
[57][70] (
Table 1). I/R includes two parts: ischemia and reperfusion. Ischemia means the sudden block of blood supply in an aerobic organ, and reperfusion means the restoration of blood supply. These processes lead to excessive production of ROS and free radicals, which result in the inflammatory cascade, cell injury, and ferroptosis
[58][71].
Table 1.
Target ferroptosis to reduce I/R injury in different organs.
| Organs |
Models and Cells |
Compounds |
Targets |
Mechanisms and
Consequences |
References |
| heart |
Sprague–Dawley rats, H9c2 cell |
Fer-1 |
ACSL4; GPX4 |
ACSL4 reduced; GPX4 increased; infarct size reduced. |
[9] |
| heart |
Mice (C57BL/6J) |
Fer-1 |
AA |
5-HETE, 11-HETE, 12-HETE, and 15-HETE reduced; infarct size reduced; left ventricular improved. |
[8] |
| heart |
Mice (C57BL/6J) |
Lip-1 |
GPX4; ROS |
GPX4 increased; ROS decreased; infarct size reduced; mitochondrial structural integrity and function maintained. |
[10] |
| renal |
HK-2 cells |
ALR |
GPX4; ROS |
ROS decreased; ALR and GPX4 colocalized; renal I/R injury protected. |
[54][67] |
| renal |
Mice |
2DG; AICAR; Fer-1 |
AMPK; PUFA |
AMPK activation; renal I/R injury protected. |
[51] |
| renal |
Mice (C57BL/6) HK-2 cells |
Panx1
deletion |
Hmox1,
NCOA4,
Fth1 |
Hmox1 upregulated, NCOA4 and FTH1 inhibited; lipid peroxidation decreased; renal I/R injury protected. |
[53][66] |
| renal |
Mice (C57BL/6) |
Fer-1 |
|
renal I/R injury protected. |
[59][72] |
| intestine |
Mice(C57BL/6) Caco-2 cells |
Lip-1; ROSI |
GPX4 ACSL4 |
GPX4 induced; ACSL4 inhibited; intestine I/R injury protected. |
[34] |
| brain |
Mice (C57BL/6) PC12 cells |
LV-shRNA-PVT1 or LV-miR-214 |
GPX4, SLC7A11 |
GPX4 and SLC7A11 increased; infarct size reduced. |
[56][69] |
| brain |
Mice (C57BL/6) |
Lip-1; Fer-1 |
|
infarct size reduced. |
[60][73] |
| brain |
Mice (Sv129/J) purified cortical neurons. |
Desferrioxamine |
HIF-1 |
HIF-1 increased; tolerance against reversible focal cerebral ischemia. |
[61][74] |
| liver |
Mice (C57BL/6) |
Lip-1 |
|
liver I/R injury reduced. |
[57][70] |
ROS are central to I/R. Inhibiting ERS, triggered by ROS, can alleviate ferroptosis in diabetes myocardial I/R injury
[9]. Compared with nondiabetic hearts, the expression of GPX4 decreases while malondialdehyde (MDA) and 4-hydroxynonenal (4-HNE) increases during myocardial I/R injury in diabetic rats. NADPH oxidase (Nox), the main donor of ROS, is responsible for the oxidative stress in diabetic rat hearts, which is closely related to AMPK
[62][75]. It is reported that the inhibition of glutaminolysis reduces the amount of ROS and increases GSH, thus inhibiting ferroptosis in heart I/R injury
[63][76]. Augmenter of liver regeneration (ALR), colocalized with GPX4, possesses the ability to inhibit ferroptosis by decreasing ROS
[54][67].
Other pathways elucidate the role of ferroptosis in I/R. Liproxstatin-1 (Lip-1), a ferroptosis inhibitor, protects heart I/R injury by reducing voltage-dependent anion channel 1 (VDAC1) levels and increasing GPX4 levels
[10][11][10,11]. Iron homeostasis is also important because a cohort study shows that the ability to rapidly process iron will be impaired during kidney I/R, which leads to ferroptosis in the end
[55][68].
Ferroptosis can also promote the neutrophil’s adhesion to coronary vascular endothelial cells by Toll-like receptors 4 (TLR4)/TIR domain-containing adapter inducing IFN-beta (Trif)/type I IFN signaling pathway, which aggravates inflammation and leads to I/R injury
[8]. Additionally, overexpression of miR-214 or silencing plasmacytoma variant 1 (PVT1) could significantly inhibit ferroptosis and reduce brain infarct size in mice
[56][69]. Renal I/R injury can be attenuated by pannexin 1 (PANX1) depletion for a decrease in mitogen-activated protein kinase (MAPK)/extracellular signal-regulated kinase (ERK) activation, which inhibits ferroptosis
[53][66].
3.2. Ferroptosis in Heart Failure
The development of heart failure is related to the loss of myocytes and is proven to have a relationship with ferroptosis. In an integrated bioinformatical analysis, Toll-like receptors 4 (TLR4) and NADPH oxidase4 (NOX4) were increased in HF. Knocking down TLR4 and NOX4 can remarkably inhibit myocyte death mediated by ferroptosis
[12]. Another mechanism revealing the role of ferroptosis in HF is mixed lineage kinase 3 (MLK3). MLK3 induces ferroptosis by regulating JUN/p53 mediated oxidative stress in chronic HF. miR-351 can inhibit this kind of heart failure by suppressing the expression of MLK3
[64][77]. Iron is critical to ferroptosis, the homeostasis of which also plays an important role in HF. Disrupting the ferritin heavy chain (Fth) gene in mice leads to HF owing to iron deposition, whereas decreasing cardiac iron in mice by lacking transferrin receptor 1 (Tfr1) also leads to HF
[65][66][78,79]. Furthermore, people suffering from HF also have decreased Tfr1 and cardiac iron
[67][63].
There are several compounds to treat HF by regulating ferroptosis. Injecting TLR4-siRNA or NOX4-siRNA lentivirus can protect the failing heart via inhibiting ferroptosis. Puerarin, an antioxidant, can inhibit lipid oxidation and iron overload in mice with heart failure induced by isoprenaline
[13]. Deferoxamine, a ferroptosis inhibitor, was reported to mitigate HF or cardiac infarction
[15]. All the above suggests that ferroptosis might be a promising target for HF.
3.3. Ferroptosis in Cardiomyopathies
3.3.1. Anticancer Drug-Induced Cardiomyopathy
Doxorubicin (DOX), a generally used anticancer drug, exerts severe side-effects, such as heart failure and cardiomyopathy. An RNA-sequencing result has shown that the expression of the heme oxygenase-1 (Hmox1) gene is significantly higher in mouse hearts treated with DOX than that of the control, which means Hmox1 is critical for DOX-induced cardiomyopathy
[15]. Moreover, the role of Hmox1 is related to ferroptosis. Nrf2 can upregulate Hmox1 to degrade heme and release free irons, thus inducing ferroptosis in cardiomyocytes
[14]. Although excess iron is hazardous, selectively cardiomyocyte Fth deficient mice also suffer mild cardiomyopathy, and dietary iron cannot rescue it but instead leads to severe heart injury through ferroptosis
[67][63]; therefore, iron homeostasis is critical to cardiomyopathy related to ferroptosis. Furthermore, knocking down P53 can increase the expression of SLC7A11 to inhibit ferroptosis and administering Fer-1 is protective for DOX-treated animals
[68][69][80,81]. Dexrazoxane (DXZ), one iron chelator, is an FDA-approved compound treating DOX-induced cardiomyopathy through inhibit ferroptosis. Mito-TEMPO, targeting mitochondria, the main organelle to produce DOX-Fe2+ induced lipid peroxide, suppresses DOX-related ferroptosis, and protects cardiomyocytes similar to Fer-1
[15][70][15,82].
3.3.2. Diabetic Cardiomyopathy
Diabetes mellitus, closely related to heart structure and function damage, can lead to cardiomyopathy via ROS’s overproduction and reduction in antioxidant ability, which is an important promoting factor of ferroptosis. NRF2, inhibiting ferroptosis by modulating antioxidant expression to reduce lipid ROS, is a promising target for diabetic cardiomyopathy (DCM)
[71][72][83,84]. Hydrogen sulfide (H2S) can increase glutathione to synthesize GSH in DCM, which reduces lipid peroxidation and further inhibits ferroptosis
[73][85]. Consequently, ferroptosis is likely to play a role in DCM.
This section may be divided by subheadings. It should provide a concise and precise description of the experimental results, their interpretation, as well as the experimental conclusions that can be drawn.
3.3.3. Iron Overload-Induced Cardiomyopathy
Iron overload (IO)-induced cardiomyopathy is thought to have a relationship with ferroptosis, which includes a high level of lipid oxidation and iron overload and usually occurs in hereditary hemochromatosis and diseases needing frequent blood transfusions such as thalassemia
[74][75][76][86,87,88]. Treating cardiomyocytes with 80 μg/mL ferric ammonium citrate for 72 h promotes eicosanoids production and arachidonic acid release, which increases lipid peroxides and affects cardiomyocyte rhythmicity, suggesting a causal link between these signals and electromechanical abnormalities in IO-induced cardiomyopathy
[77][89]. Mitochondrial Ca uniporter (mCU), which mediates iron uptake, is important to IO-induced cardiomyopathy. Compared to those of mCU+/+ (WT) mice, mCU−/− (KO) have lower mitochondrial iron and ROS levels and better systolic function; therefore, ferroptosis participates in IO-induced cardiomyopathy. Administering ferrostatin-1 protects heart function also confirms this point of view
[78][90].
3.4. Ferroptosis in Atherosclerosis
Atherosclerosis is mainly caused by lipid deposition, endothelial cell damage, the proliferation of VSMC, and transformation of macrophages, and so on
[79][54]. All these processes involve ferroptosis. Lipid deposition, especially PUFAs, is the substrate of the LOX or Fenton reaction, leading to oxidized lipids and inducing ferroptosis. In high-fat diet (HFD)-fed Apo E-/-mice, the administering of Fer-1, a ferroptosis inhibitor, can promote the expressions of SLC7A11 and GPX4, partially inhibit the iron accumulation and lipid peroxidation, as well as increase ox-LDL-induced mouse aortic endothelial cells (MAECs) viability
[16]. Moreover, endothelial cells can be damaged by lipid ROS, increasing the endothelial permeability, promoting lipid deposition, and initiating atherosclerosis. In HUVECs, miRNA17-92 overexpression greatly alleviates erastin-induced cell death through the A20-ACSL4 axis
[1]. That means inhibiting ferroptosis in endothelial cells can alleviate endothelial damage and reduce the probability of atherosclerosis. Studies showed that the iron level in healthy arterial tissue is less than that in atherosclerotic plaque
[80][91]. Iron overload is also a key point to induce ferroptosis; therefore, restricting iron, especially nontransferrin bound iron (NTBI), a deleterious iron form, is thought to be therapeutic for atherosclerosis by inhibiting ferroptosis
[79][81][54,92]. Furthermore, iron overload even changes macrophage function, leading it to polarize toward the proinflammatory subtype
[82][58]. This chronic inflammation of the macrophage contributes to the formation of atherosclerotic necrotic core and destabilization of plaque
[83][93]; therefore, ferroptosis can induce atherosclerosis via iron overload and macrophage changes.
3.5. Ferroptosis in Aging
Ferroptosis is related to aging because of the role of iron and excess ROS. Iron accumulates with aging as the need for iron decreases as the metabolic rate reduces, and the amount of hemoglobin also decreases with aging
[84][94]. One experiment on rabbits shows that, compared to that of young ones, aged rabbits possess more free iron, released from ferritin, and this leads to further oxidative damage, which may be related to ferroptosis, because the ferroptosis inhibitor, deferoxamine, is protective of this kind of damage
[85][95].
3.6. Ferroptosis in Vascular or Ventricular Remodeling
The homeostasis of iron is critical to ferroptosis and the pathogenesis of vascular and ventricular remodeling. One study shows that iron deficiency contributes to vascular remodeling in the pulmonary of rat
[86][96]. On the contrary, another study reports that restricting iron may moderately alleviate hypoxia-induced vascular remodeling in the pulmonary of mice
[87][97]. As for the heart, it is believed that the myocardial iron in the infarct zone is adverse for ventricular remodeling
[88][98]; therefore, the role of iron and ferroptosis in vascular remodeling needs more investigation (
Figure 2).
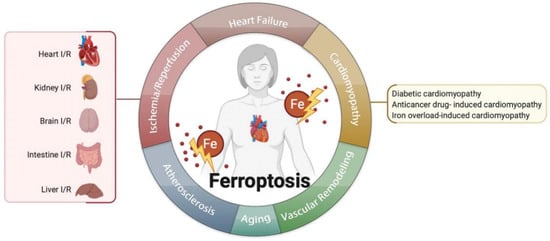
Figure 2. Ferroptosis is related to multiple cardiovascular diseases, such as ischemia/reperfusion, heart failure, cardiomyopathies, atherosclerosis, aging, vascular remodeling, and so on.图 2. 铁死亡与多种心血管疾病有关,如缺血/再灌注、心力衰竭、心肌病、动脉粥样硬化、衰老、血管重塑等。