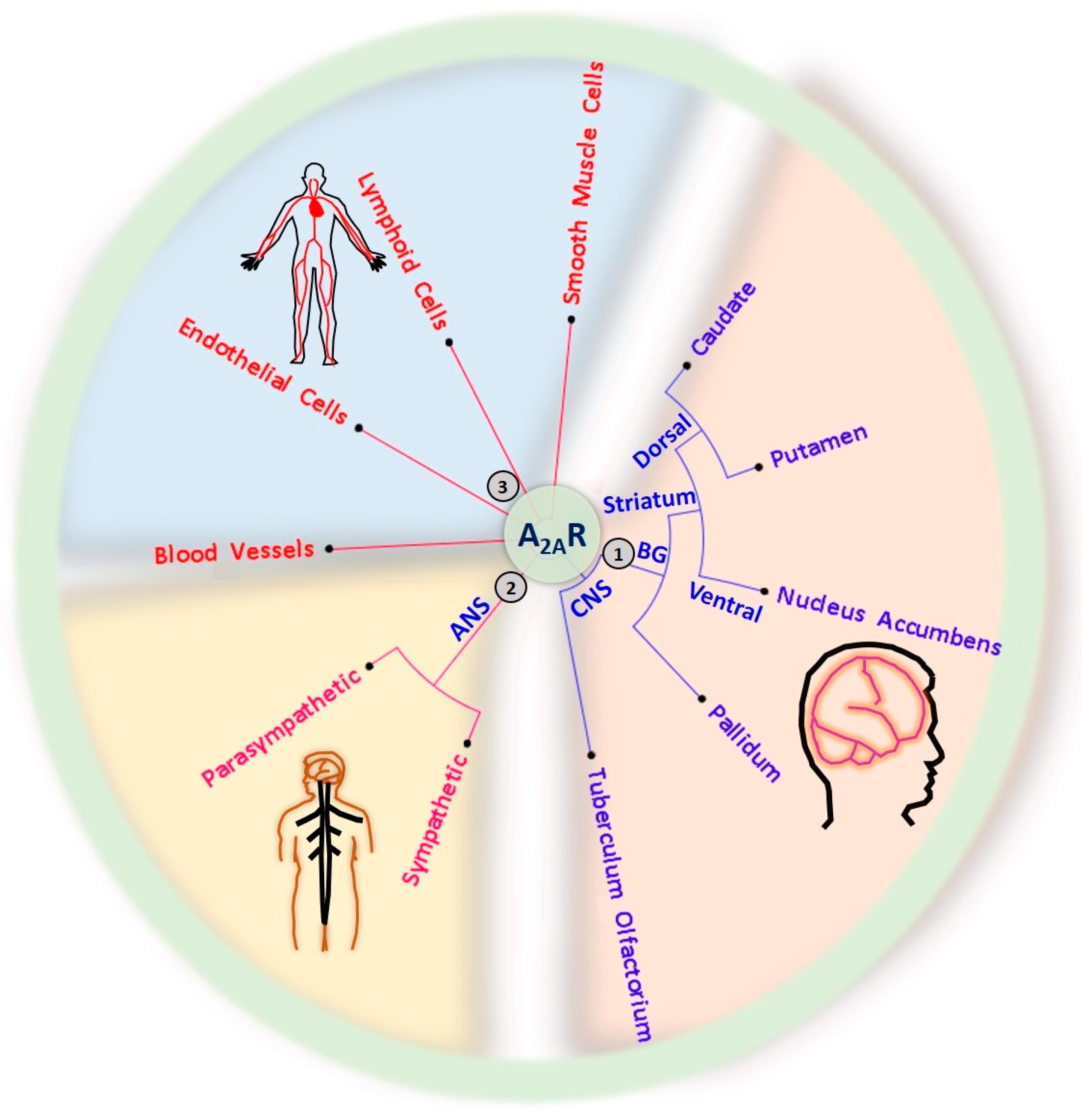
Adenosine is a naturally occurring purine nucleoside that regulates various physiologic functions, including inflammation and wound healing, cardiac contraction, blood vessel formation, vasodilation, learning, memory, sleep, and arousal. Adenosine is released by neurons and glial cells. Extracellular adenosine modulates neuronal excitability, synaptic plasticity, and the release and reuptake of several neurotransmitters. The effects of extracellular adenosine are modulated via four subtypes of G-protein coupled adenosine receptors (GPCRs), denoted A1, A2A, A2B, and A3. Adenosine A2A receptors (A2ARs) are broadly expressed in the brain, cardiovascular system, blood vessels, spleen, thymus, leukocytes, and lung, making them an important drug target. The therapeutic potential of targeting adenosine A2ARs is immense due to their broad expression in the body and central nervous system. The role of A2ARs in cardiovascular function, inflammation, sleep/wake behaviors, cognition, and other primary nervous system functions has been extensively studied.
- adenosine A2A receptors
- allosteric modulator
- insomnia
- slow-wave sleep
- inflammation
- cardiovascular function
- body temperature
- drug development
1. Introduction
2. Adenosine and Its Receptors
3. A2AR and Its Physiologic Roles
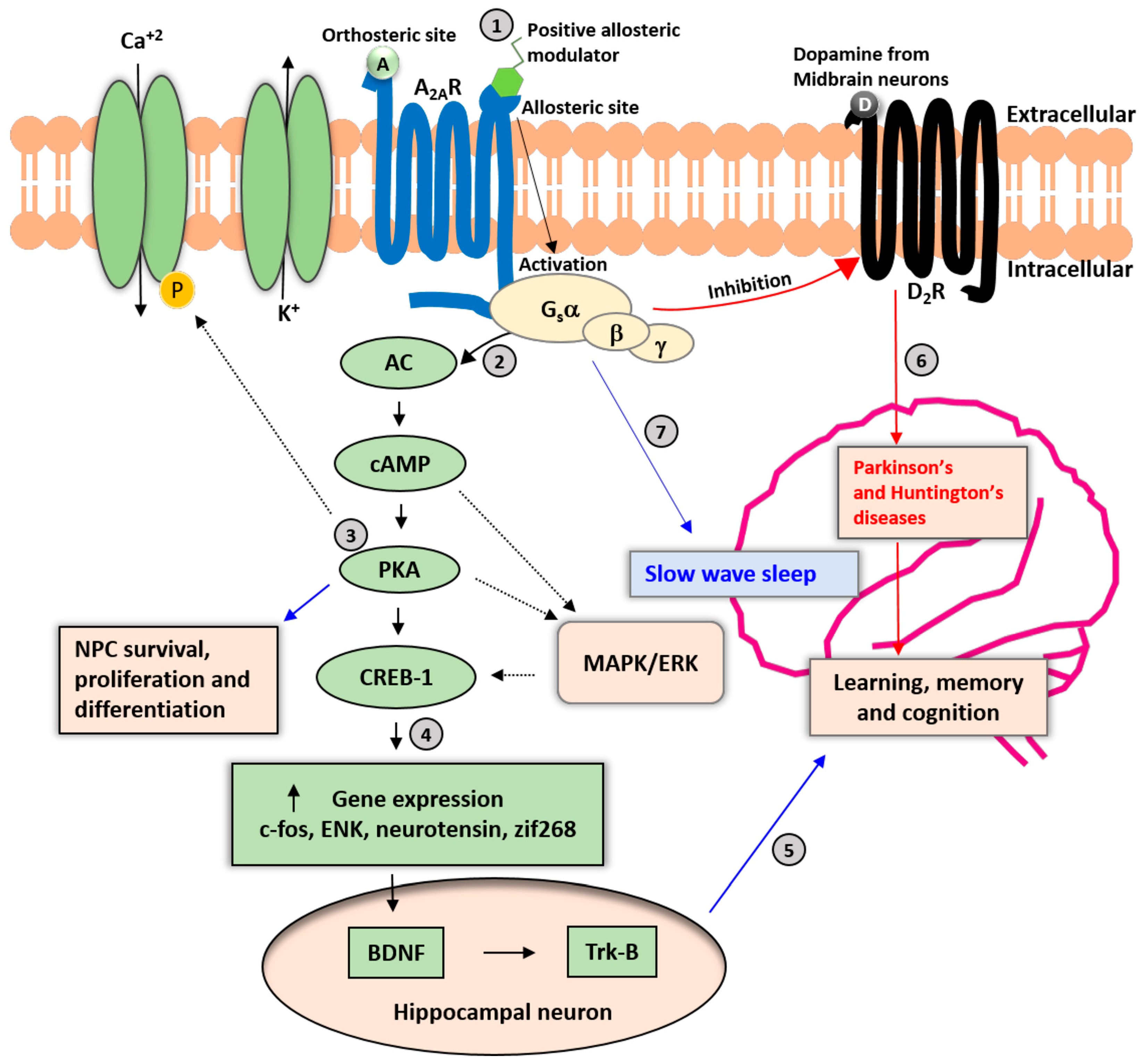
4. The Concept of Allosteric Modulation
5. Allosteric A2AR Modulation
6. Allosteric A2AR Modulators and Their Potential Clinical Application
|
Name |
Type |
Pharmacology |
Structure |
Physiologic Effects |
|---|---|---|---|---|
|
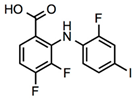
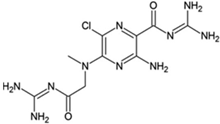
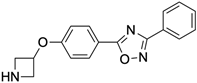
3,4-Difluoro-2-((2-fluoro-4-iodophenyl)amino)benzoic acid |
Allosteric enhancer/modulator |
Enhanced adenosine signaling at mouse A2ARs. |
|
Induced slow wave sleep without affecting cardiovascular function or body temperature in wild-type male mice [94,95][66][67]. |
|
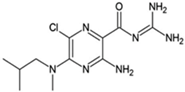
AEA061 |
Allosteric enhancer/modulator |
Enhanced adenosine and inosine signaling and increased effect of the A2AR agonist CGS 21680. |
Not disclosed |
Inhibited the production of tumor necrosis factor-α, macrophage inflammatory protein-1α, 1β, and 2, interleukin-1α, keratinocyte chemokine, and RANTES (regulated upon activation, normal T cell expressed and presumably secreted) in macrophages and splenocytes, reduced circulating plasma tumor necrosis factor-α and monocyte chemoattractant protein-1 levels, and increased plasma interleukin-10 during lipopolysaccharide-induced endotoxemia [96,97][68][69]. |
|
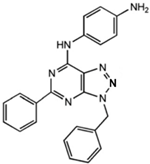
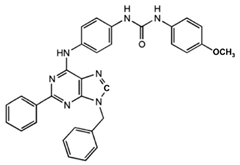
N-(3-Benzyl-5-phenyl-3H-[1,2,3]triazolo[4,5-d]- pyrimidin-7yl-)-(4-aminophenyl)-amine |
Allosteric modulator |
Inhibited the binding of antagonists and agonists at the A2AR orthosteric site [93][65]. |
|
Unknown |
|
N6-[(4-Nitro)-phenyl]-9-benzyl-2-phenyladenine |
Allosteric modulator |
Inhibited the binding of antagonists and agonists at the A2AR orthosteric site [93][65]. |
|
Unknown |
|
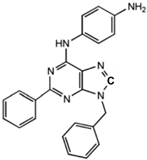
N6-[(4-Amino)-phenyl]-9-benzyl-2-phenyladenine |
Allosteric modulator |
Inhibited the binding of antagonists and agonists at the A2AR orthosteric site [93][65]. |
|
Unknown |
|
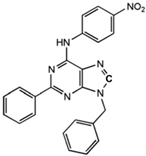
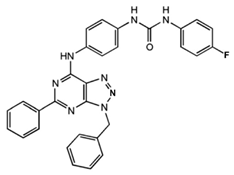
1-[4-(3-Benzyl-5-phenyl-3H-[1,2,3]triazolo[4,5-d]-pyrimidin-7-ylamino)-phenyl]-3-(4-fluorophenyl)-urea |
Allosteric modulator |
Modulated the binding of antagonist and agonist at the A2AR orthosteric site [93][65]. |
|
Unknown |
|
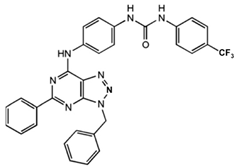
1-[4-(3-Benzyl-5-phenyl-3H-[1,2,3]triazolo[4,5-d]-pyrimidin-7-ylamino)-phenyl]-3-(4-trifluoromethylphenyl)- urea |
Allosteric modulator |
Modulated the binding of antagonist and agonist at the A2AR orthosteric site [93][65]. |
|
Unknown |
|
1-[4-(9-Benzyl-2-phenyl-9H-purin-6-ylamino)- phenyl]-3-(4-methoxyphenyl-urea |
Allosteric modulator |
Modulated the binding of antagonist and agonist at the A2AR orthosteric site [93][65]. |
|
Unknown |
|
Amiloride |
Allosteric modulator |
Increased the dissociation rate of the antagonist ZM-241,385 at rat A2ARs [18,71][54][57]. |
|
Unknown |
|
Benzamil |
Allosteric modulator |
Increased the dissociation rate of the antagonist ZM-241,385 at rat A2ARs [71][57]. |
|
Unknown |
|
HMA; 5-(N,N-hexamethylene)amiloride |
Allosteric modulator |
Increased the dissociation rate of the antagonist ZM-241,385 at rat A2ARs [71][57]. |
|
Unknown |
|
MGCMA; 5-(N-methyl-N-guanidinocarbonyl-methyl)amiloride |
Allosteric modulator |
Increased the dissociation rate of the antagonist ZM-241,385 at rat A2ARs [71][57]. |
|
Unknown |
|
MIBA; 5-(N-methyl-N-isobutyl)amiloride |
Allosteric modulator |
Increased the dissociation rate of the antagonist ZM-241,385 at rat A2ARs [71][57]. |
|
Unknown |
|
Phenamil |
Allosteric modulator |
Increased the dissociation rate of the antagonist ZM-241,385 at rat A2ARs [71][57]. |
|
Unknown |
|
Sodium Ion |
Allosteric modulator |
Na+ |
Unknown |
|
|
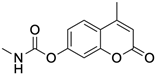
PD120918 {4-methyl-7-[(methyl- amino)carbonyl]oxy}-2H-1-benzopyran-2-one} |
Allosteric modulator |
Enhanced agonist radioligand binding to rat striatal A2ARs without functional enhancement [18,91][54][63]. |
|
Unknown |
|
Fg754 |
Allosteric modulator |
Increased the dissociation rate of the agonist CGS21680 at A2ARs expressing HEK-293 cells [72][58]. |
|
Unknown |
|
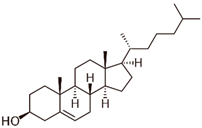
Cholesterol |
Allosteric modulator |
Decreased the dissociation rate of the agonist NECA at A2ARs-embedded nanodiscs [73][59]. |
|
Unknown |
References
- Adair, T.H. Growth Regulation of the Vascular System: An Emerging Role for Adenosine. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2005, 289, R283–R296.
- Chen, J.-F. Adenosine Receptor Control of Cognition in Normal and Disease. Int. Rev. Neurobiol. 2014, 119, 257–307.
- Feoktistov, I.; Biaggioni, I.; Cronstein, B.N. Adenosine Receptors in Wound Healing, Fibrosis and Angiogenesis. Handb. Exp. Pharmacol. 2009, 383–397.
- Headrick, J.P.; Ashton, K.J.; Rose’meyer, R.B.; Peart, J.N. Cardiovascular Adenosine Receptors: Expression, Actions and Interactions. Pharmacol. Ther. 2013, 140, 92–111.
- Hein, T.W.; Wang, W.; Zoghi, B.; Muthuchamy, M.; Kuo, L. Functional and Molecular Characterization of Receptor Subtypes Mediating Coronary Microvascular Dilation to Adenosine. J. Mol. Cell. Cardiol. 2001, 33, 271–282.
- Lazarus, M.; Chen, J.-F.; Huang, Z.-L.; Urade, Y.; Fredholm, B.B. Adenosine and Sleep. Handb. Exp. Pharmacol. 2019, 253, 359–381.
- Ohta, A.; Sitkovsky, M. Role of G-Protein-Coupled Adenosine Receptors in Downregulation of Inflammation and Protection from Tissue Damage. Nature 2001, 414, 916–920.
- Zhou, X.; Oishi, Y.; Cherasse, Y.; Korkutata, M.; Fujii, S.; Lee, C.-Y.; Lazarus, M. Extracellular Adenosine and Slow-Wave Sleep Are Increased after Ablation of Nucleus Accumbens Core Astrocytes and Neurons in Mice. Neurochem. Int. 2019, 124, 256–263.
- Chin, J.H. Adenosine Receptors in Brain: Neuromodulation and Role in Epilepsy. Ann. Neurol. 1989, 26, 695–698.
- Ciruela, F.; Casadó, V.; Rodrigues, R.J.; Luján, R.; Burgueño, J.; Canals, M.; Borycz, J.; Rebola, N.; Goldberg, S.R.; Mallol, J.; et al. Presynaptic Control of Striatal Glutamatergic Neurotransmission by Adenosine A1-A2A Receptor Heteromers. J. Neurosci. 2006, 26, 2080–2087.
- Kamikubo, Y.; Shimomura, T.; Fujita, Y.; Tabata, T.; Kashiyama, T.; Sakurai, T.; Fukurotani, K.; Kano, M. Functional Cooperation of Metabotropic Adenosine and Glutamate Receptors Regulates Postsynaptic Plasticity in the Cerebellum. J. Neurosci. 2013, 33, 18661–18671.
- Matos, M.; Augusto, E.; Santos-Rodrigues, A.D.; Schwarzschild, M.A.; Chen, J.-F.; Cunha, R.A.; Agostinho, P. Adenosine A2A Receptors Modulate Glutamate Uptake in Cultured Astrocytes and Gliosomes. Glia 2012, 60, 702–716.
- Fredholm, B.B.; Ijzerman, A.P.; Jacobson, K.A.; Linden, J.; Müller, C.E. International Union of Basic and Clinical Pharmacology. LXXXI. Nomenclature and Classification of Adenosine Receptors—An Update. Pharmacol. Rev. 2011, 63, 1–34.
- Fredholm, B.B.; Ijzerman, A.P.; Jacobson, K.A.; Klotz, K.N.; Linden, J. International Union of Pharmacology. XXV. Nomenclature and Classification of Adenosine Receptors. Pharmacol. Rev. 2001, 53, 527–552.
- Drury, A.N.; Szent-Györgyi, A. The Physiological Activity of Adenine Compounds with Especial Reference to their Action upon the Mammalian Heart. J. Physiol. 1929, 68, 213–237.
- Haskó, G.; Pacher, P.; Deitch, E.A.; Vizi, E.S. Shaping of Monocyte and Macrophage Function by Adenosine Receptors. Pharmacol. Ther. 2007, 113, 264–275.
- Sattin, A.; Rall, T.W. The Effect of Adenosine and Adenine Nucleotides on the Cyclic Adenosine 3′, 5′-Phosphate Content of Guinea Pig Cerebral Cortex Slices. Mol. Pharmacol. 1970, 6, 13–23.
- Fredholm, B.B. Adenosine, an Endogenous Distress Signal, Modulates Tissue Damage and Repair. Cell Death Differ. 2007, 14, 1315–1323.
- Schrader, J. Metabolism of Adenosine and Sites of Production in the Heart. In Regulatory Function of Adenosine, Proceedings of the International Symposium on Adenosine, Charlottesville, Virginia, 7–11 June 1982; Berne, R.M., Rall, T.W., Rubio, R., Eds.; Developments in Pharmacology 2; Springer: Boston, MA, USA, 1983; pp. 133–156. ISBN 978-1-4613-3909-0.
- Ballarín, M.; Fredholm, B.B.; Ambrosio, S.; Mahy, N. Extracellular Levels of Adenosine and its Metabolites in the Striatum of Awake Rats: Inhibition of Uptake and Metabolism. Acta Physiol. Scand. 1991, 142, 97–103.
- Boison, D.; Singer, P.; Shen, H.-Y.; Feldon, J.; Yee, B.K. Adenosine Hypothesis of Schizophrenia—Opportunities for Pharmacotherapy. Neuropharmacology 2012, 62, 1527–1543.
- Cacciari, B.; Pastorin, G.; Bolcato, C.; Spalluto, G.; Bacilieri, M.; Moro, S. A2B Adenosine Receptor Antagonists: Recent Developments. Mini Rev. Med. Chem. 2005, 5, 1053–1060.
- Burnstock, G.; Cocks, T.; Crowe, R.; Kasakov, L. Purinergic Innervation of the Guinea-Pig Urinary Bladder. Br. J. Pharmacol. 1978, 63, 125–138.
- Matsumoto, T.; Tostes, R.C.; Webb, R.C. Alterations in Vasoconstrictor Responses to the Endothelium-Derived Contracting Factor Uridine Adenosine Tetraphosphate Are Region Specific in DOCA-Salt Hypertensive Rats. Pharmacol. Res. 2012, 65, 81–90.
- Göblyös, A.; Ijzerman, A.P. Allosteric Modulation of Adenosine Receptors. Purinergic Signal. 2009, 5, 51–61.
- Fredholm, B.B.; Chen, J.-F.; Cunha, R.A.; Svenningsson, P.; Vaugeois, J.-M. Adenosine and Brain Function. Int. Rev. Neurobiol. 2005, 63, 191–270.
- Maenhaut, C.; Van Sande, J.; Libert, F.; Abramowicz, M.; Parmentier, M.; Vanderhaegen, J.J.; Dumont, J.E.; Vassart, G.; Schiffmann, S. RDC8 Codes for an Adenosine A2 Receptor with Physiological Constitutive Activity. Biochem. Biophys. Res. Commun. 1990, 173, 1169–1178.
- Chern, Y.; King, K.; Lai, H.L.; Lai, H.T. Molecular Cloning of a Novel Adenosine Receptor Gene from Rat Brain. Biochem. Biophys. Res. Commun. 1992, 185, 304–309.
- Furlong, T.J.; Pierce, K.D.; Selbie, L.A.; Shine, J. Molecular Characterization of a Human Brain Adenosine A2 Receptor. Brain Res. Mol. Brain Res. 1992, 15, 62–66.
- Ledent, C.; Vaugeois, J.M.; Schiffmann, S.N.; Pedrazzini, T.; El Yacoubi, M.; Vanderhaeghen, J.J.; Costentin, J.; Heath, J.K.; Vassart, G.; Parmentier, M. Aggressiveness, Hypoalgesia and High Blood Pressure in Mice Lacking the Adenosine A2a Receptor. Nature 1997, 388, 674–678.
- Meng, F.; Xie, G.X.; Chalmers, D.; Morgan, C.; Watson, S.J.; Akil, H. Cloning and Expression of the A2a Adenosine Receptor from Guinea Pig Brain. Neurochem. Res. 1994, 19, 613–621.
- de Lera Ruiz, M.; Lim, Y.-H.; Zheng, J. Adenosine A2A Receptor as a Drug Discovery Target. J. Med. Chem. 2014, 57, 3623–3650.
- Kull, B.; Svenningsson, P.; Fredholm, B.B. Adenosine A2A Receptors Are Colocalized with and Activate Golf in Rat Striatum. Mol. Pharmacol. 2000, 58, 771–777.
- Ritter, J.; Flower, R.; Henderson, G.; Loke, Y.K.; MacEwan, D.; Rang, H. Rang & Dale’s Pharmacology, 9th ed.; Elsevier: Amsterdam, The Netherlands, 2018.
- Neubig, R.R.; Spedding, M.; Kenakin, T.; Christopoulos, A. International Union of Pharmacology Committee on Receptor Nomenclature and Drug Classification International Union of Pharmacology Committee on Receptor Nomenclature and Drug Classification. XXXVIII. Update on Terms and Symbols in Quantitative Pharmacology. Pharmacol. Rev. 2003, 55, 597–606.
- Hu, J. Allosteric Modulators of the Human Calcium-Sensing Receptor: Structures, Sites of Action, and Therapeutic Potentials. Endocr. Metab. Immune Disord. Drug Targets 2008, 8, 192–197.
- Kenakin, T. Pharmacology in Drug Discovery and Development, 2nd ed.; Elsevier: Amsterdam, The Netherlands, 2016.
- Koshland, D.E.; Némethy, G.; Filmer, D. Comparison of Experimental Binding Data and Theoretical Models in Proteins Containing Subunits. Biochemistry 1966, 5, 365–385.
- Monod, J.; Wyman, J.; Changeux, J.-P. On the Nature of Allosteric Transitions: A Plausible Model. J. Mol. Biol. 1965, 12, 88–118.
- Monod, J.; Changeux, J.-P.; Jacob, F. Allosteric Proteins and Cellular Control Systems. J. Mol. Biol. 1963, 6, 306–329.
- Estébanez-Perpiñá, E.; Arnold, L.A.; Nguyen, P.; Rodrigues, E.D.; Mar, E.; Bateman, R.; Pallai, P.; Shokat, K.M.; Baxter, J.D.; Guy, R.K.; et al. A Surface on the Androgen Receptor That Allosterically Regulates Coactivator Binding. Proc. Natl. Acad. Sci. USA 2007, 104, 16074–16079.
- Hughes, T.S.; Giri, P.K.; de Vera, I.M.S.; Marciano, D.P.; Kuruvilla, D.S.; Shin, Y.; Blayo, A.-L.; Kamenecka, T.M.; Burris, T.P.; Griffin, P.R.; et al. An Alternate Binding Site for PPARγ Ligands. Nat. Commun. 2014, 5, 3571.
- Bono, F.; De Smet, F.; Herbert, C.; De Bock, K.; Georgiadou, M.; Fons, P.; Tjwa, M.; Alcouffe, C.; Ny, A.; Bianciotto, M.; et al. Inhibition of Tumor Angiogenesis and Growth by a Small-Molecule Multi-FGF Receptor Blocker with Allosteric Properties. Cancer Cell 2013, 23, 477–488.
- De Smet, F.; Christopoulos, A.; Carmeliet, P. Allosteric Targeting of Receptor Tyrosine Kinases. Nat. Biotechnol. 2014, 32, 1113–1120.
- Catterall, W.A.; Cestèle, S.; Yarov-Yarovoy, V.; Yu, F.H.; Konoki, K.; Scheuer, T. Voltage-Gated Ion Channels and Gating Modifier Toxins. Toxicon 2007, 49, 124–141.
- Olsen, R.W.; Chang, C.-S.S.; Li, G.; Hanchar, H.J.; Wallner, M. Fishing for Allosteric Sites on GABA(A) Receptors. Biochem. Pharmacol. 2004, 68, 1675–1684.
- Spedding, M.; Kenny, B.; Chatelain, P. New Drug Binding Sites in Ca2+ Channels. Trends Pharmacol. Sci. 1995, 16, 139–142.
- Taly, A.; Corringer, P.-J.; Guedin, D.; Lestage, P.; Changeux, J.-P. Nicotinic Receptors: Allosteric Transitions and Therapeutic Targets in the Nervous System. Nat. Rev. Drug Discov. 2009, 8, 733–750.
- Traynelis, S.F.; Wollmuth, L.P.; McBain, C.J.; Menniti, F.S.; Vance, K.M.; Ogden, K.K.; Hansen, K.B.; Yuan, H.; Myers, S.J.; Dingledine, R. Glutamate Receptor Ion Channels: Structure, Regulation, and Function. Pharmacol. Rev. 2010, 62, 405–496.
- Colquhoun, D. Binding, Gating, Affinity and Efficacy: The Interpretation of Structure-Activity Relationships for Agonists and of the Effects of Mutating Receptors. Br. J. Pharmacol. 1998, 125, 924–947.
- Fenton, A.W. Allostery: An Illustrated Definition for the “Second Secret of Life”. Trends Biochem. Sci. 2008, 33, 420–425.
- Nussinov, R.; Tsai, C.-J. Allostery in Disease and in Drug Discovery. Cell 2013, 153, 293–305.
- Christopoulos, A.; Changeux, J.-P.; Catterall, W.A.; Fabbro, D.; Burris, T.P.; Cidlowski, J.A.; Olsen, R.W.; Peters, J.A.; Neubig, R.R.; Pin, J.-P.; et al. International Union of Basic and Clinical Pharmacology. XC. Multisite Pharmacology: Recommendations for the Nomenclature of Receptor Allosterism and Allosteric Ligands. Pharmacol. Rev. 2014, 66, 918–947.
- Gao, Z.-G.; Kim, S.-K.; Ijzerman, A.P.; Jacobson, K.A. Allosteric Modulation of the Adenosine Family of Receptors. Mini Rev. Med. Chem. 2005, 5, 545–553.
- Jacobson, K.A.; Gao, Z.-G. Adenosine Receptors as Therapeutic Targets. Nat. Rev. Drug Discov. 2006, 5, 247–264.
- Gao, Z.-G.; Melman, N.; Erdmann, A.; Kim, S.G.; Müller, C.E.; IJzerman, A.P.; Jacobson, K.A. Differential Allosteric Modulation by Amiloride Analogues of Agonist and Antagonist Binding at A(1) and A(3) Adenosine Receptors. Biochem. Pharmacol. 2003, 65, 525–534.
- Gao, Z.G.; Ijzerman, A.P. Allosteric Modulation of A(2A) Adenosine Receptors by Amiloride Analogues and Sodium Ions. Biochem. Pharmacol. 2000, 60, 669–676.
- Lu, Y.; Liu, H.; Yang, D.; Zhong, L.; Xin, Y.; Zhao, S.; Wang, M.-W.; Zhou, Q.; Shui, W. Affinity Mass Spectrometry-Based Fragment Screening Identified a New Negative Allosteric Modulator of the Adenosine A2A Receptor Targeting the Sodium Ion Pocket. ACS Chem. Biol. 2021, 16, 991–1002.
- Huang, S.K.; Almurad, O.; Pejana, R.J.; Morrison, Z.A.; Pandey, A.; Picard, L.-P.; Nitz, M.; Sljoka, A.; Prosser, R.S. Allosteric Modulation of the Adenosine A2A Receptor by Cholesterol. eLife 2022, 11, e73901.
- Christopoulos, A. Allosteric Binding Sites on Cell-Surface Receptors: Novel Targets for Drug Discovery. Nat. Rev. Drug Discov. 2002, 1, 198–210.
- Christopoulos, A. Advances in G Protein-Coupled Receptor Allostery: From Function to Structure. Mol. Pharmacol. 2014, 86, 463–478.
- Jiang, Q.; Lee, B.X.; Glashofer, M.; van Rhee, A.M.; Jacobson, K.A. Mutagenesis Reveals Structure-Activity Parallels between Human A2A Adenosine Receptors and Biogenic Amine G Protein-Coupled Receptors. J. Med. Chem. 1997, 40, 2588–2595.
- Bruns, R.F.; Fergus, J.H. Allosteric Enhancement of Adenosine A1 Receptor Binding and Function by 2-Amino-3-Benzoylthiophenes. Mol. Pharmacol. 1990, 38, 939–949.
- Göblyös, A.; de Vries, H.; Brussee, J.; Ijzerman, A.P. Synthesis and Biological Evaluation of a New Series of 2,3,5-Substituted -Thiadiazoles as Modulators of Adenosine A1 Receptors and Their Molecular Mechanism of Action. J. Med. Chem. 2005, 48, 1145–1151.
- Giorgi, I.; Biagi, G.; Bianucci, A.M.; Borghini, A.; Livi, O.; Leonardi, M.; Pietra, D.; Calderone, V.; Martelli, A. N6-1,3-Diphenylurea Derivatives of 2-Phenyl-9-Benzyladenines and 8-Azaadenines: Synthesis and Biological Evaluation as Allosteric Modulators of A2A Adenosine Receptors. Eur. J. Med. Chem. 2008, 43, 1639–1647.
- Korkutata, M.; Saitoh, T.; Cherasse, Y.; Ioka, S.; Duo, F.; Qin, R.; Murakoshi, N.; Fujii, S.; Zhou, X.; Sugiyama, F.; et al. Enhancing Endogenous Adenosine A2A Receptor Signaling Induces Slow-Wave Sleep without Affecting Body Temperature and Cardiovascular Function. Neuropharmacology 2019, 144, 122–132.
- Korkutata, M.; Saitoh, T.; Feng, D.; Murakoshi, N.; Sugiyama, F.; Cherasse, Y.; Nagase, H.; Lazarus, M. Allosteric Modulation of Adenosine A2A Receptors in Mice Induces Slow-Wave Sleep without Cardiovascular Effects. Sleep Med. 2017, 40, e181.
- Welihinda, A.A.; Amento, E.P. Positive Allosteric Modulation of the Adenosine A2a Receptor Attenuates Inflammation. J. Inflamm. 2014, 11, 37.
- Welihinda, A.A.; Kaur, M.; Raveendran, K.S.; Amento, E.P. Enhancement of Inosine-Mediated A2AR Signaling through Positive Allosteric Modulation. Cell. Signal. 2018, 42, 227–235.