Insulin-like growth factor-1 (IGF-1) and its binding proteins and receptors are widely expressed in the central nervous system (CNS), proposing IGF-1-induced neurotrophic actions in normal growth, development, and maintenance. However, while there is convincing evidence that the IGF-1 system has specific endocrine roles in the CNS, the concept is emerging that IGF-I might be also important in disorders such as ischemic stroke, brain trauma, Alzheimer’s disease, epilepsy, etc., by inducing neuroprotective effects towards glutamate-mediated excitotoxic signaling pathways. Research in rodent models has demonstrated rescue of pathophysiological and behavioral abnormalities when IGF-1 was administered by different routes, and several clinical studies have shown safety and promise of efficacy in neurological disorders of the CNS.
- glutamate-mediated excitotoxicity
- signaling pathways
- insulin-like growth factor-1
- neuroprotection
- animal models
- clinical trials
1. Introduction
2. IGF-1 Modulation of Glutamate-Induced Synaptic Plasticity
3. IGF-1 Modulates Calcium Pathway
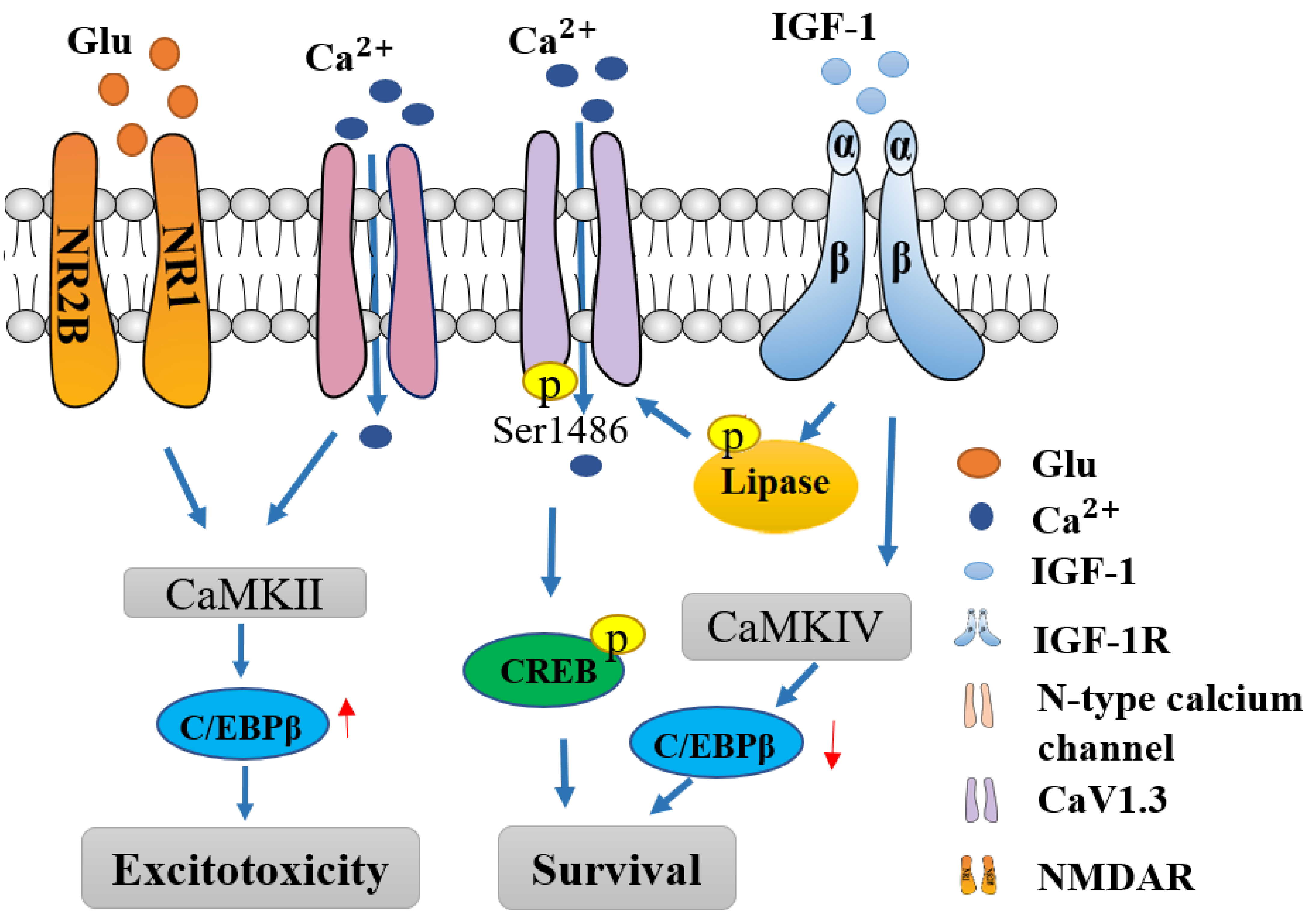
4. IGF-I Confers Neuroprotection towards Neurological Diseases with Glutamate Excitotoxicity
| Disease | Animal Models | Human Patients | Reference |
|---|---|---|---|
| Ischemic Stroke | Attenuated infarct size with IGF-1 treatment in MCAO and improved post-stroke neurological behaviors. | Inverse correlation between circulating IGF-1 levels and stroke incidence; The levels of IGF-1in the serum is also inversely associated with the neurological deficits following stroke. | [5][27] |
| Traumatic brain injury (TBI) | IGF-1 is neuroprotective. Functional neurological improvement of motor and cognitive functions in different TBI models. | IGF-1 clinical trials in TBI demonstrate that IGF-1 administration either alone or in combination with GH was safe to humans and successful in improving metabolic parameters in moderate-to-severe TBI patients. | [48] |
| Amyotrophic Lateral Sclerosis (ALS) | In mouse models of ALS rhIGF-1 delayed disease onset, reduced muscle atrophy, promoted peripheral motor nerve regeneration, and extended life. | Randomized, double-blind, placebo-controlled, phase two and three clinical trials reaffirmed that rhIGF-1 administration was safe and well tolerated in most subjects but efficacy was not statistically significant. | [26] |
| Alzheimer’s Disease (AD) | In mice with increased cerebral beta-amyloid plaques serum IGF-1 modulated brain levels of beta-amyloid and prevented premature death | Multicenter, cross-sectional study to assess the relationship between IGF-1 and cognitive decline indicated that serum IGF-IGFBP-3 levels were implicated in men with AD. However, a double-blind, multicenter study using growth hormone secretagogue MK-677 which stimulates upregulation and circulation of IGF-1, failed to show efficacy in slowing disease progression. | [25][28][29][30] |
| Autism spectrum disorder (ASD)- Phelan-McDermid Syndrome (PMS) | I.p. injection of rhIGF-1 in Shank3-deficient mice at clinically approved doses of 0.24 mg/kg/day for 2 weeks reversed the electro-physiological deficits and demonstrated reduced AMPAR-mediated transmission and showed normal LTP comparable to the wild type control mice |
A clinical trial using 0.24 mg/kg/day of rhIGF-1 in divided doses, in nine children with PMS (Shank3 deficient) demonstrated safety, tolerability, and efficacy. | [20][31][33] |
| ASD- Fragile X Syndrome (FXS) | In Fmr1 knockout mice characterized by reduced excitatory synaptic currents, enhanced glutamate receptor dependent-LTD, 100 mg/kg i.p. injection of IGF-1 analog Trofinetide (NNZ-2566) resulted with reduced hyperactivity, improved LSTM and LTP, and normalized social recognition and behaviors. | Phase II randomized, double-blind, placebo-controlled, parallel-group, confirmed the safety, tolerability and efficacy at the high dose of treatment with oral administration of Trofinetide at 35 or 70 mg/kg twice daily, in 72 adolescent or adult males with FXS. | [35][36] |
| Friedreich’s ataxia (FRDA) | IGF-I in FRDA-like transgenic mice (YG8R mice) conferred neuroprotection and normalized motor coordination. | In a clinical proof of concept pilot study, patients were treated s.c. with IGF-1 therapy with 50 μg/kg twice a day for 12 months and tolerability and decrease in the progression of neurological symptoms was measured, together with long-term stability of cardiac function. | [37][38][39] |
| Huntington’s disease (HD) | IGF-1 intranasal delivery rescues HD phenotype in YAC128 mice. | In 219 patients with genetically documented HD and in 71 sex- and age-matched controls, IGF-1 serum levels were significantly higher in patients than in controls, indicating somatotropic axis is overactive to confer neuroprotection. | [40][41] |
| Epilepsy | IGF-I ameliorated hippocampal neurodegeneration and protected against cognitive deficits in an animal model of temporal lobe epilepsy. | 57 patients with focal epilepsy and 35 healthy controls were evaluated for IGF-1 level; reduced serum levels of IGF-1were found to correlate with age and cardiovagal function, a parameter of cerebral autoregulation (the breath-hold index). Patients with a longer history of epilepsy, presented higher seizure frequency, and temporal lobe epilepsy and had lower serum levels of IGF-1. | [42][43] |
References
- Lewitt, M.S.; Boyd, G.W. The Role of Insulin-Like Growth Factors and Insulin-Like Growth Factor-Binding Proteins in the Nervous System. Biochem. Insights 2019, 12, 1178626419842176.
- Yagami, T.; Yamamoto, Y.; Koma, H. Pathophysiological Roles of Intracellular Proteases in Neuronal Development and Neurological Diseases. Mol. Neurobiol. 2019, 56, 3090–3112.
- Mehta, A.; Prabhakar, M.; Kumar, P.; Deshmukh, R.; Sharma, P.L. Excitotoxicity: Bridge to various triggers in neurodegenerative disorders. Eur J. Pharmacol. 2013, 698, 6–18.
- Olloquequi, J.; Cornejo-Cordova, E.; Verdaguer, E.; Soriano, F.X.; Binvignat, O.; Auladell, C.; Camins, A. Excitotoxicity in the pathogenesis of neurological and psychiatric disorders: Therapeutic implications. J. Psychopharmacol. 2018, 32, 265–275.
- Hayes, C.A.; Valcarcel-Ares, M.N.; Ashpole, N.M. Preclinical and clinical evidence of IGF-1 as a prognostic marker and acute intervention with ischemic stroke. J. Cereb. Blood. Flow. Metab. 2021, 41, 2475–2491.
- Zheng, P.; Tong, W. IGF-1: An endogenous link between traumatic brain injury and Alzheimer disease? J. Neurosurg. Sci. 2017, 61, 416–421.
- Carro, E.; Trejo, J.L.; Nunez, A.; Torres-Aleman, I. Brain repair and neuroprotection by serum insulin-like growth factor I. Mol. Neurobiol. 2003, 27, 153–162.
- Nanou, E.; Catterall, W.A. Calcium Channels, Synaptic Plasticity, and Neuropsychiatric Disease. Neuron. 2018, 98, 466–481.
- Magee, J.C.; Grienberger, C. Synaptic Plasticity Forms and Functions. Annu. Rev. Neurosci. 2020, 43, 95–117.
- Aberg, N.D.; Brywe, K.G.; Isgaard, J. Aspects of growth hormone and insulin-like growth factor-I related to neuroprotection, regeneration, and functional plasticity in the adult brain. Science 2006, 6, 53–80.
- Chin, P.C.; Majdzadeh, N.; D’Mello, S.R. Inhibition of GSK3beta is a common event in neuroprotection by different survival factors. Brain Res. Mol. Brain Res. 2005, 137, 193–201.
- Peineau, S.; Taghibiglou, C.; Bradley, C.; Wong, T.P.; Liu, L.; Lu, J.; Lo, E.; Wu, D.; Saule, E.; Bouschet, T.; et al. LTP inhibits LTD in the hippocampus via regulation of GSK3beta. Neuron 2007, 53, 703–717.
- Ding, Q.; Vaynman, S.; Akhavan, M.; Ying, Z.; Gomez-Pinilla, F. Insulin-like growth factor I interfaces with brain-derived neurotrophic factor-mediated synaptic plasticity to modulate aspects of exercise-induced cognitive function. Neuroscience 2006, 140, 823–833.
- Marshall, J.; Dolan, B.M.; Garcia, E.P.; Sathe, S.; Tang, X.; Mao, Z.; Blair, L.A. Calcium channel and NMDA receptor activities differentially regulate nuclear C/EBPbeta levels to control neuronal survival. Neuron 2003, 39, 625–639.
- Ogundele, O.M.; Ebenezer, P.J.; Lee, C.C.; Francis, J. Stress-altered synaptic plasticity and DAMP signaling in the hippocampus-PFC axis; elucidating the significance of IGF-1/IGF-1R/CaMKIIalpha expression in neural changes associated with a prolonged exposure therapy. Neuroscience 2017, 353, 147–165.
- Ramsey, M.M.; Weiner, J.L.; Moore, T.P.; Carter, C.S.; Sonntag, W.E. Growth hormone treatment attenuates age-related changes in hippocampal short-term plasticity and spatial learning. Neuroscience 2004, 129, 119–127.
- Calamandrei, G.; Alleva, E. Neuronal growth factors, neurotrophins and memory deficiency. Behav. Brain Res. 1995, 66, 129–132.
- Ceprian, M.; Fulton, D. Glial Cell AMPA Receptors in Nervous System Health, Injury and Disease. Int. J. Mol. Sci. 2019, 20.
- Sivakumar, V.; Ling, E.A.; Lu, J.; Kaur, C. Role of glutamate and its receptors and insulin-like growth factors in hypoxia induced periventricular white matter injury. Glia 2010, 58, 507–523.
- Bozdagi, O.; Tavassoli, T.; Buxbaum, J.D. Insulin-like growth factor-1 rescues synaptic and motor deficits in a mouse model of autism and developmental delay. Mol. Autism 2013, 4, 9.
- Moutin, E.; Sakkaki, S.; Compan, V.; Bouquier, N.; Giona, F.; Areias, J.; Goyet, E.; Hemonnot-Girard, A.L.; Seube, V.; Glasson, B.; et al. Restoring glutamate receptosome dynamics at synapses rescues autism-like deficits in Shank3-deficient mice. Mol. Psychiatry 2021.
- Blair, L.A.; Marshall, J. IGF-1 modulates N and L calcium channels in a PI 3-kinase-dependent manner. Neuron 1997, 19, 421–429.
- Gao, L.; Blair, L.A.; Salinas, G.D.; Needleman, L.A.; Marshall, J. Insulin-like growth factor-1 modulation of CaV1.3 calcium channels depends on Ca2+ release from IP3-sensitive stores and calcium/calmodulin kinase II phosphorylation of the alpha1 subunit EF hand. J. Neurosci. 2006, 26, 6259–6268.
- Sanchez, J.C.; Lopez-Zapata, D.F.; Francis, L.; De Los Reyes, L. Effects of estradiol and IGF-1 on the sodium calcium exchanger in rat cultured cortical neurons. Cell. Mol. Neurobiol. 2011, 31, 619–627.
- Russo, V.C.; Gluckman, P.D.; Feldman, E.L.; Werther, G.A. The insulin-like growth factor system and its pleiotropic functions in brain. Endocr. Rev. 2005, 26, 916–943.
- Costales, J.; Kolevzon, A. The therapeutic potential of insulin-like growth factor-1 in central nervous system disorders. Neurosci. Biobehav. Rev. 2016, 63, 207–222.
- Serhan, A.; Boddeke, E.; Kooijman, R. Insulin-Like Growth Factor-1 Is Neuroprotective in Aged Rats With Ischemic Stroke. Front. Aging Neurosci. 2019, 11, 349.
- Sevigny, J.J.; Ryan, J.M.; van Dyck, C.H.; Peng, Y.; Lines, C.R.; Nessly, M.L.; Group, M.K.P.S. Growth hormone secretagogue MK-677: No clinical effect on AD progression in a randomized trial. Neurology 2008, 71, 1702–1708.
- Duron, E.; Funalot, B.; Brunel, N.; Coste, J.; Quinquis, L.; Viollet, C.; Belmin, J.; Jouanny, P.; Pasquier, F.; Treluyer, J.M.; et al. Insulin-like growth factor-I and insulin-like growth factor binding protein-3 in Alzheimer’s disease. J. Clin. Endocrinol. Metab. 2012, 97, 4673–4681.
- Ostrowski, P.P.; Barszczyk, A.; Forstenpointner, J.; Zheng, W.; Feng, Z.P. Meta-Analysis of Serum Insulin-Like Growth Factor 1 in Alzheimer’s Disease. PLoS ONE 2016, 11, e0155733.
- Frank, Y. The Neurological Manifestations of Phelan-McDermid Syndrome. Pediatr. Neurol. 2021, 122, 59–64.
- Deacon, R.M.; Glass, L.; Snape, M.; Hurley, M.J.; Altimiras, F.J.; Biekofsky, R.R.; Cogram, P. NNZ-2566, a novel analog of (1-3) IGF-1, as a potential therapeutic agent for fragile X syndrome. Neuromolecular Med. 2015, 17, 71–82.
- Kolevzon, A.; Bush, L.; Wang, A.T.; Halpern, D.; Frank, Y.; Grodberg, D.; Rapaport, R.; Tavassoli, T.; Chaplin, W.; Soorya, L.; et al. A pilot controlled trial of insulin-like growth factor-1 in children with Phelan-McDermid syndrome. Mol. Autism 2014, 5, 54.
- Berry-Kravis, E.; Horrigan, J.P.; Tartaglia, N.; Hagerman, R.; Kolevzon, A.; Erickson, C.A.; Hatti, S.; Snape, M.; Yaroshinsky, A.; Stoms, G.; et al. A Double-Blind, Randomized, Placebo-Controlled Clinical Study of Trofinetide in the Treatment of Fragile X Syndrome. Pediatr. Neurol. 2020, 110, 30–41.
- Castro, J.; Garcia, R.I.; Kwok, S.; Banerjee, A.; Petravicz, J.; Woodson, J.; Mellios, N.; Tropea, D.; Sur, M. Functional recovery with recombinant human IGF1 treatment in a mouse model of Rett Syndrome. Proc. Natl. Acad. Sci. USA 2014, 111, 9941–9946.
- Glaze, D.G.; Neul, J.L.; Percy, A.; Feyma, T.; Beisang, A.; Yaroshinsky, A.; Stoms, G.; Zuchero, D.; Horrigan, J.; Glass, L.; et al. A Double-Blind, Randomized, Placebo-Controlled Clinical Study of Trofinetide in the Treatment of Rett Syndrome. Pediatr. Neurol. 2017, 76, 37–46.
- Franco, C.; Fernandez, S.; Torres-Aleman, I. Frataxin deficiency unveils cell-context dependent actions of insulin-like growth factor I on neurons. Mol. Neurodegener. 2012, 7, 51.
- Franco, C.; Genis, L.; Navarro, J.A.; Perez-Domper, P.; Fernandez, A.M.; Schneuwly, S.; Torres Aleman, I. A role for astrocytes in cerebellar deficits in frataxin deficiency: Protection by insulin-like growth factor I. Mol. Cell. Neurosci. 2017, 80, 100–110.
- Sanz-Gallego, I.; Torres-Aleman, I.; Arpa, J. IGF-1 in Friedreich’s Ataxia–proof-of-concept trial. Cerebellum Ataxias 2014, 1, 10.
- Lopes, C.; Ribeiro, M.; Duarte, A.I.; Humbert, S.; Saudou, F.; Pereira de Almeida, L.; Hayden, M.; Rego, A.C. IGF-1 intranasal administration rescues Huntington’s disease phenotypes in YAC128 mice. Mol. Neurobiol. 2014, 49, 1126–1142.
- Saleh, N.; Moutereau, S.; Durr, A.; Krystkowiak, P.; Azulay, J.P.; Tranchant, C.; Broussolle, E.; Morin, F.; Bachoud-Levi, A.C.; Maison, P. Neuroendocrine disturbances in Huntington’s disease. PLoS ONE 2009, 4, e4962.
- Miltiadous, P.; Stamatakis, A.; Koutsoudaki, P.N.; Tiniakos, D.G.; Stylianopoulou, F. IGF-I ameliorates hippocampal neurodegeneration and protects against cognitive deficits in an animal model of temporal lobe epilepsy. Exp. Neurol. 2011, 231, 223–235.
- Chen, S.F.; Jou, S.B.; Chen, N.C.; Chuang, H.Y.; Huang, C.R.; Tsai, M.H.; Tan, T.Y.; Tsai, W.C.; Chang, C.C.; Chuang, Y.C. Serum Levels of Brain-Derived Neurotrophic Factor and Insulin-Like Growth Factor 1 Are Associated With Autonomic Dysfunction and Impaired Cerebral Autoregulation in Patients With Epilepsy. Front. Neurol. 2018, 9, 969.
- Lu, X.C.; Si, Y.; Williams, A.J.; Hartings, J.A.; Gryder, D.; Tortella, F.C. NNZ-2566, a glypromate analog, attenuates brain ischemia-induced non-convulsive seizures in rats. J. Cereb. Blood Flow Metab. 2009, 29, 1924–1932.
- Kamato, D.; Mitra, P.; Davis, F.; Osman, N.; Chaplin, R.; Cabot, P.J.; Afroz, R.; Thomas, W.; Zheng, W.; Kaur, H.; et al. Gaq proteins: Molecular pharmacology and therapeutic potential. Cell. Mol. Life Sci. 2017, 74, 1379–1390.
- Frater, J.; Lie, D.; Bartlett, P.; McGrath, J.J. Insulin-like Growth Factor 1 (IGF-1) as a marker of cognitive decline in normal ageing: A review. Ageing Res. Rev. 2018, 42, 14–27.
- Green, C.J.; Holly, J.M.; Bayer, A.; Fish, M.; Ebrahim, S.; Gallacher, J.; Ben-Shlomo, Y. The role of IGF-I, IGF-II, and IGFBP-3 in male cognitive aging and dementia risk: The Caerphilly Prospective Study. J. Alzheimers Dis. 2014, 41, 867–875.
- Martin, E.D.; Sanchez-Perez, A.; Trejo, J.L.; Martin-Aldana, J.A.; Cano Jaimez, M.; Pons, S.; Acosta Umanzor, C.; Menes, L.; White, M.F.; Burks, D.J. IRS-2 Deficiency impairs NMDA receptor-dependent long-term potentiation. Cereb. Cortex 2012, 22, 1717–1727.