Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Chengjun Wu and Version 3 by Conner Chen.
Alternative splicing of pre-mRNA is a key mechanism for increasing the complexity of proteins in humans, causing a diversity of expression of transcriptomes and proteomes in a tissue-specific manner. Alternative splicing is an essential process in post-transcriptional mRNA processing, and produces various mature mRNAs with different structures and functions.
- alternative splicing
- splicing factors
- diseases
- drugs
1. Introduction
Alternative splicing is an essential process in post-transcriptional mRNA processing, and produces various mature mRNAs with different structures and functions. In this process, exons are taken together in different combinations and introns are removed. Recent data indicate that each transcript of protein-coding genes contain 11 exons and produce 5.4 mRNAs on average [1]. So far, seven basic types of alternative splicing have been identified, including exon skipping, alternative 5′-splice site, alternative 3′-splice site, mutually exclusive exons, intron retention, alternative promoter, and alternative polyadenylation [2] (Figure 1). A notable example of alternative splicing is the human gene TTN which encodes muscle protein titin and contains 364 coding-exons and 4039 different splicing events which have been identified by RNA-sequencing [3]. Most genes generate at least two transcript variants. The alternative spliced mRNAs are further translated into many protein variants which differ in function and structure. The precision and diversity of alternative splicing events are aided by many significant factors, such as the strength or weakness of splice sites, the concentration and combination of enhancing and silencing splicing factors, chromatin modifications, and RNA secondary architectures [3]. Since the activity of splicing factors and the spliced variants change the developing process of diseases, they can also serve as experimental indicators or biomarkers for diagnosis.
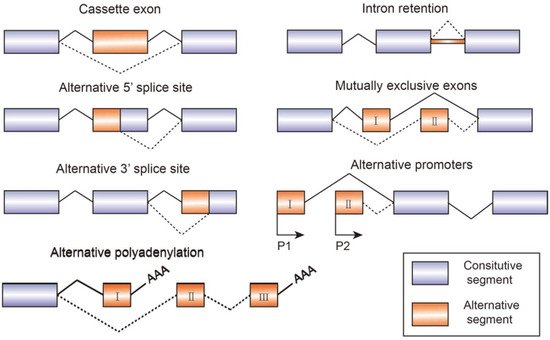
Figure 1. Schematic figure representing seven modes of alternative splicing of pre-mRNA: cassette exon, intron retention, alternative 5′ splice site, mutually exclusive exons, alternative 3′ splice site, alternative promoters, and alternative polyadenylation. Alternative splicing sites are connected by dashed lines. Boxes represent exons and lines represent introns.
1.1. Function and Assembly of Spliceosome
The spliceosome recruited by cis-acting elements and trans-acting factors plays a critical role in regulating the constitutive and alternative splicing procedure. It is an enormous macromolecular complex containing five small nuclear RNAs (U1, U2, U4, U5, and U6), and hundreds of combined proteins called small nuclear ribonucleoproteins (snRNPs). The complex splicing regulation process is carried out by virtue of dynamic assembles of snRNPs in a stepwise fashion. Here we show a brief overview. Firstly, U1 snRNP binds to 5′-ss GU di-nucleotide, splicing factor 1 (SF1) and U2AF65 bind to branch point site (BPS) and the polypyrimidine tract (PPT), respectively, forming the E complex. Secondly, U2 snRNP base-pairing interacts with the BPS displacing SF1 to form complex A. Afterwards, it recruits U4/U6/U5 tri-snRNP, leading to the formation of complex B, in which U5 snRNP binds to 3′-ss and U6 snRNP binds to U2 snRNP. Meanwhile, U1 and U4 snRNP are released. Complex C is then formed. Followed by two transesterification steps, intron is folded into a lariat and 5′-ss is cleaved. Finally, the two exons are linked together and the lariat is released. snRNPs can be used for recycling [4]. It has been indicated that mutations of splicing factors can disrupt the expression ratios of small nuclear RNAs and spliceosome assembly, inducing premature pathogenic termination of mRNA translation [5].
1.2. Essential Structures and Elements for Alternative Splicing Regulation
The boundaries between exons and introns are illustrated by a 5′-splice site (5′-ss) with highly conserved GU nucleotide combination, a 3′-splice site (3′-ss) with highly conserved AG nucleotide sequence, a branch-point sequence (BPS) located at 18–40 nucleotides upstream of the 3′-ss, and a polypyrimidine tract (PPT) which is also critical in recognizing 3′-ss. The decisions of removal or retention toward specific exons depend on the role of cis-acting elements, which are short nucleoside sequences containing RNA binding sites located on the pre-mRNA. The cis-acting elements include exonic splicing enhancers (ESEs), exonic splicing silencers (ESSs), intronic splicing enhancers (ISEs), and intronic splicing silencers (ISSs) [6]. The two major families of cellular RNA binding proteins participating in the splicing process are serine/arginine-rich (SR) protein and the heterogeneous nuclear ribonucleoproteins (hnRNPs). The structures of RNA recognition motif (RRM) and serine/arginine-rich domain (RS domain) are what make SR proteins functional in splicing regularly. For instance, SR proteins mediate the interaction between U1 snRNP and 5′-ss and recruit U2 snRNP to the 3′-ss. Besides, they often cooperate with other positive splicing factors to form enhancing complexes, such as TRA2, SRRM1, and SRRM2 [7]. However, SR proteins and hnRNPs families generally have an opposite effect during choosing the alternative splice sites and exons by definition act in a competitive manner. ESEs and ISEs mainly recruit SR proteins acting as splicing activators, while ESSs and ISSs are usually recognized by hnRNPs proteins function as splicing repressors [8]. For example, splicing of exon 6B from β-tropomyosin gene depends on the G-rich intronic sequence (S3) and downstream of exon 6B, which can act as either an enhancer or a silencer. ASF/SF2 and SC35 bind to S3 and positively stimulate the splicing of exon 6B, whereas hnRNP A1 competitively disrupts their interaction [9]. Mutation of spliceosome components and dysregulation of splicing mechanisms can affect the network of downstream splicing targets, causing human diseases such as cancer, neurodegenerative disorders, and metabolic diseases [10].
1.3. Tissue Specificity of Alternative Splicing
The distribution of alternative splicing factors is tissue-specific, which is the reason for the diversity of cell differentiation and tissue specificity. It has been indicated that more than 50% of genes express different alternative spliced isoforms among tissues. The human brain, the most functionally diverse tissue, contains several specific splicing factors, including nPTB, NOVA1, and NOVA2. In the process of neuronal differentiation, the expression of splicing factors shifted from PTB to nPTB. The upregulation of PTB is responsible for approximately a quarter of nervous system-specific alternative splicing. The CELF family of proteins containing three RRMs is broadly expressed in many tissues. Among them CELF1, CELF2, CELF5, and CELF6 are located in the brain, serving as alternative splicing regulators which mainly target gene TNTT2. In addition, CELF2 and CELF5 are also distributed in the heart and skeletal muscle tissues. RBM35a and RBM35b are epithelial cells-specific splicing factors, controlling the expression of epithelial characteristics related exons [7].
2. Alternative Splicing Factors and Spliced Isoforms in Relation to Cancer Pathogenesis
Cancer is a complex disease which can be caused by various factors. It has been determined that gene expression profiles of tumor cells are different from normal samples. Instable cellular homeostasis is an important cause of cancer, and has been reported to be closely related to aberrant alternative splicing. Since alternative splicing plays a key role in post-transcriptional regulation and controls the formation of spliced variants, the mutations and changed level of splice factors may contribute to tumorigenesis.
Abnormal expressions of specific splicing isoforms can cause specific cancers but also can serve as a biomarkers and therapeutic targets. The TCGA database allows one to analyze the expression of alternative splicing patterns during cancer progression [11]. Increasing evidence demonstrates the dysregulation of alternative splicing leads to the production of tumor-associated isoforms that further impact cellular activity [4], such as sustaining proliferation, preventing cell death, rewiring cell metabolism, promoting angiogenesis, enabling cell invasion and metastatic dissemination, and enabling drug resistance [12]. For example, the mutation of SF3B1 is highly related with several cancers including chronic lymphocytic leukemia (CLL), cutaneous melanomas, and uveal melanomas. SF3B1 contains an essential region which interacts with SF3B14a and further forms a complex with U2AF2, playing a key role in BPS recognition. However, the mutation of SF3B1 at exons 12–15 disturbs the interaction between SF3B1 and SF3B14a, leading to the prevention of BPS recognition and 3′-ss mis-selection. Moreover, the mutated SF3B1 functions as an anti-apoptotic factor which is regularly detected in various cancers in order to sustain the cell proliferation [13]. SR proteins are responsible for multiple cellular physiological process and involved in the alteration of gene expression in tumors. SRSF1 is often upregulated in breast tumors through binding with MYC, leading to increasing of cell proliferation and decreasing of cell apoptosis. Additionally, SRSF1 overexpression in lung cancer leads to resistance to the chemotherapy drugs cisplatin and topotecan. Previous studies have demonstrated that the SRSF2 mutant is associated with myelodysplastic syndromes (MDS), since the mutated SRSF2 expression alters the binding specificity, inducing the inclusion of a premature termination codon (PTC). This PTC locates in EZH2 region which encoding a histone methyltransferase related to the pathogenesis of MDS. SRSF6 overexpression is detected in skin cancer, promoting the splicing of cassette exons and inducing hyperplasia [14]. The hnRNPs play an important role in regulation of pre-mRNA splicing. Abnormal expression of hnRNPs affects RNA splicing, results in alteration of RNA expression levels, and further causes the occurrence of cancer. As the most abundant hnRNPs, hnRNP A1/A2 has been reported to be upregulated in lung cancer. Overexpression of hnRNP A1/A2 may function as carcinogenic factor to promote cell proliferation. Moreover, hnRNP A1 and hnRNP A2 participate in recognizing and protecting telomeric sequences. Thus, they are related to cancer regulation. In contrary, silencing of hnRNPA1/A2 expression causes tumor cell apoptosis [15]. In addition, miRNAs can modulate splicing-factor expression and function as oncogenes. miR-30a-5p and miR-181a-5p regulate SRSF7 in renal tumors, thereby altering the splicing pattern [16][17][16,17]. Overexpression of splicing associated miRNAs has been detected in a variety of cancers. For example, repression of SRSF1 results in upregulation of miR-10a and miR-10b, thereby promoting terminal differentiation of neuroblastoma cells.
Alternative splicing events generate protein isoforms related to cancer hallmarks, promoting tumorigenesis. Gene RPSkKB1 alternatively encode two protein isoforms, full-length RPS6KB1-1 and RPS6KB1-2 lacking kinase domain. The production of RPS6KB1-2 is regulated by SRSF1 mediated AS, and contributes to tumor growth in lung and breast cancer, while RPS6KB1-1 suppresses proliferation of cancer cells [15][18][15,18]. Similarly, oncogene CCND1 encoding cyclin D1 has two isoforms, namely conventional cyclin D1a and cyclin D1b lacking the C-terminal protein domains. Cyclin D1b C-terminal domain encoded by the exon 5 is a GSK-3β phosphorylation site, allowing the cyclin D1b to be transported from the nucleus to the cytoplasm. Altered selection of 5′-ss induce exclusion of exon 5, thereby causing cyclin D1b to be trapped in the nucleus. As opposed to cyclin D1a, the overexpression of cyclin D1b variant was observed in breast cancer tissues compared to normal breast tissues, which characterizes metastasis and invasive migration of cancer cells mediated by αvβ3 and toll-like receptor 4 (TLR4) [19][20][21][22][23][19,20,21,22,23] (Figure 2).
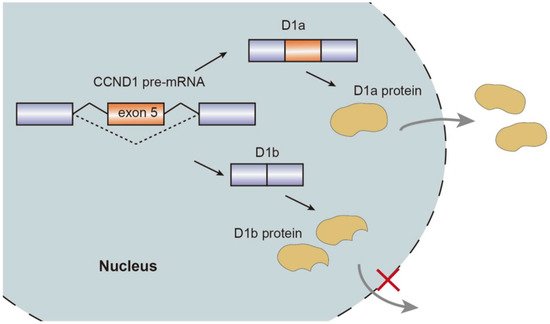
Figure 2.
Schematic representation of CCND1 pre-mRNA splicing mechanism, isoforms, and nuclear transport.
3. The Relationship between Alternative Splicing and Neurological Diseases
Neurodegenerative disease constitutes a variety of mental and neuromuscular disorders, including Alzheimer’s disease (AD), Parkinson’s disease (PD), spinal muscular atrophy (SMA), and familial dysautonomia (FD). It has been found that alternative splicing plays an important role in neurological diseases.
Neuro-oncological ventral antigens 1 and 2 (NOVA1 and NOVA2) are two members of the NOVA family, regulating neuron-specific alternative splicing. They belong to splicing factors and mainly enriched in brain tissue and are responsible for neuronal viability and maturity. NOVA1 is distributed in the cerebellum and spine, whereas NOVA2 is located in the cortex. NOVAs regulates various receptors and voltage-gated channels which are critical for the signal transduction of neuromuscular junctions. As an alternative splicing method for a key regulator of glycine α2 exon 3A (GlyRα2E3A) pre-mRNA splicing, NOVA1 recognizes an adjacent intronic splice site, alternatively inducing exon 3A inclusion. NOVA1, together with TDP-43, participates in RNA recognition and induces various neurodegenerative diseases such as schizophrenia and amyotrophic lateral sclerosis [24]. NOVA1 participates in the splicing of RNA-binding protein RBM8A and regulates RNA metabolism, causing neurological damage-like symptoms of AD.
Alternative splicing events frequently occur in the progression of AD. FynT is an isoform of tyrosine kinase protein Fyn, which is upregulated in AD patients and correlated with chronic neuro-inflammation [25]. Alternative splicing of exon 15 of Amyloid precursor protein (APP) transcript changes the ratio of APP proteolytic fragments betaA4 and p3, which contributes to the formation of amyloid plaque and aggravate AD [26]. Another major toxic in AD is microtubule-stabilizing protein tau. Tau has two isoforms, four microtubule repeats (4R-tau), and three microtubule repeats (3R-tau). The generation of both isoforms are determined by the inclusion or skipping of exon 10, which are further regulated by splicing factor SRSF6 [27]. The two isoforms expression level of 3R-tau and 4R-tau have to maintain a balance in healthy individuals. However, dysregulation of tau splicing disturbs the ratios, thereby causing neurofibrillary degeneration in AD patients [28]. AMPA serves as a critical mediator of synaptic transmission. AMPA receptor subunits of PD patients have been regulated alternatively, which are potential biomarkers for PD diagnosis and therapies [29]. SMA is a common autosomal recessive disease which is partially caused by the decrease of SMN protein. Human beings encoded two SMN genes, namely SMN1 and SMN2. They are almost identical and encode SMN protein, which is crucial for snRNP assembly. However, SMN2 is vulnerable to mutate in exon 7 due to the replacement of C by T at position 6. Hence, during the post transcriptional processing, exon 7 is skipped in SMN2 pre-mRNA splicing, generating truncated isoforms which cannot compensate the function of SMN [30][31][30,31]. Sodium butyrate has been detected to modulate exon 7 inclusion of SMN2 transcript, thereby producing the full-length SMN protein [32]. Familial dysautonomia (FD) is a recessive disease mainly caused by the mutation of the i-kappa-B kinase complex associated protein (IKBKAP). The mutated IKBKAP loses its function, and thereby FD patients show a demyelination phenotype. Single-base changes in less well-conserved nucleotides affecting the selection of splice site are responsible for numerous genetic diseases. In this case, the mutation of nucleotide T to C occurring at position 6 of intron 20 disrupts the interaction between IKBKAP pre-mRNA and U1 snRNA, and weakens the splicing from the intronic 5′-ss. Additionally, computational analyses identified that upstream 3′-ss also contains weak splicing signals, further causing the defining of exon 20 almost impossible. [33] (Figure 3).
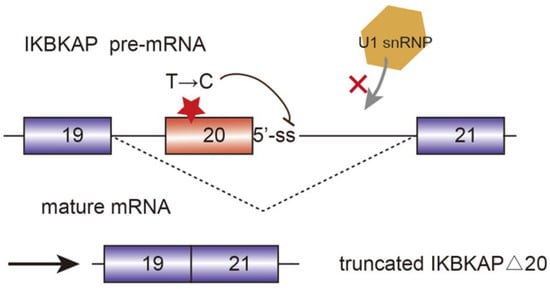
Figure 3. Mutated i-kappa-B kinase complex associated protein (IKBKAP) plays a role in the development of familial dysautonomia (FD). The single nucleotide mutation from T to C disrupts the interaction between pre-mRNA and U1 snRNP and induce the exon 20 exclusion.
This entry is adapted from 10.3390/genes13030401