Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Asma Amamou and Version 2 by Peter Tang.
Intestinal fibrosis is a common complication in inflammatory bowel disease (IBD) without specific treatment. Macrophages are the key actors in inflammatory responses and the wound healing process. By their exceptional ability to integrate diverse stimuli in their surrounding environment, macrophages display a multitude of phenotypes to underpin a broad spectrum of functions, from the initiation to the resolution of inflammation following injury. The hypothesis that distinct macrophage subtypes could be involved in fibrogenesis and wound healing is emerging and could open up new therapeutic perspectives in the treatment of intestinal fibrosis.
- inflammatory bowel diseases
- fibrosis
- macrophages
1. Introduction
Inflammatory bowel diseases (IBD) are chronic relapsing and remitting inflammatory diseases affecting the gastrointestinal tract, resulting from complex interactions between genes, environment exposure, especially food, the immune system, and gut microbiota. IBD are often progressive and lifelong pathologies leading to irreversible intestinal damage causing disability and morbidity in patients [1]. Among these complications, intestinal fibrosis is the most common. Indeed, intestinal fibrosis affects up to 50% of Crohn’s disease patients (CD) and to a lesser degree up to 11% of ulcerative colitis (UC) patients [1]. Intestinal fibrosis is characterised by exaggerated scar tissue formation in intestinal mucosa resulting from chronic inflammation. In IBD, while specific targeted therapies have made a major impact on inflammation, the incidence of fibrostenotic complications has not substantially reduced and the development of new therapies is urgently needed [2]. About 80% of patients undergo surgical resection with loss of viable tissue. [3][4][3,4].
2. Macrophages as Key Players in Fibrogenesis
The gut mucosa hosts the largest population of macrophages in our organism. These intestinal macrophages play pivotal roles in the maintenance of intestinal homeostasis. Although macrophages are effector cells of the innate immune system, they are also involved in mucosal healing, angiogenesis, and tissue metabolism [5][6]. Macrophages are present in all the phases of mucosal healing exhibiting distinct phenotypes associated with specific tasks [6][7]. Commonly, macrophages, referred to as pro-inflammatory phenotype M1, are the first macrophages in place following an injury to clean the local environment of a wound from bacteria, dead cells, and debris [7][8]. During the initiation of tissue repair, macrophages undergo phenotype change into anti-inflammatory and pro-regenerative macrophages, referred to as M2, which produce key mediators, such as transforming growth factor β1 (TGF-β1), to promote the migration and differentiation of tissue fibroblasts which produce extracellular matrix (ECM) components that facilitate wound healing [6][7]. Finally, during the remodelling phase, macrophages release matrix metalloproteinases (MMP), restoring tissue integrity [8][9]. Thus, wound healing is a complex, tightly regulated process; however, when injuries are repeated, this process persists and becomes uncontrolled, leading to complications, especially fibrosis [9][10]. The functional plasticity of macrophages depends on adaptation to their tissue environment, changes which provoke reprogramming of their metabolism and the induction of a complex gene program regulated by numerous transcription factor families [10][11]. The composition of the local tissue microenvironment, such as cytokines, growth factors, microbial products, along with other immune and stromal cells, are critical determinants of the macrophage responses. Over the last decade it has become clear that macrophage function in tissue repair is complex and contributes to several chronic diseases [11][12]. Considering their critical role in the inflammatory response and mucosal healing, macrophages have generated considerable interest in deciphering their involvement in chronic inflammatory disorders, including IBD [12][13][13,14]. IBD patients and experimental models of colitis show an accumulation of macrophages in inflamed tissue [14][15][15,16]. It is now well established that M1 macrophages play a critical role in the chronicity of inflammation associated with IBD [14][15]. Interestingly, many recent studies have demonstrated the involvement of macrophages in fibrogenesis of the lungs, kidneys, and pancreas [16][17][18][17,18,19]. The contribution of macrophages to intestinal fibrogenesis remains poorly studied; however, because similarity in histomorphology is observed in fibrosis involving all organs, some of the mechanisms underpinning fibrogenesis are likely to be common to all these organs, including in the gastrointestinal tract.
In the lungs, liver, and kidneys, a co-localization of macrophages and myofibroblasts has been observed at fibrotic sites [19][20][21][20,21,22]. These studies highlight the importance of the interaction between macrophages and myofibroblasts in the fibrotic tissue. Proliferation and differentiation of myofibroblasts are the central events in the fibrogenesis process [22][23]. While multiple factors influence the promotion of myofibroblast expansion, TGF-β1 remains the most critical in fibrosis progression [23][24]. TGF-β1 mediates progressive intestinal fibrosis in both experimental and human IBD through Smad2/Smad3 pathway activation by stimulating ECM production whilst inhibiting its degradation. Moreover, TGF-β1 acts also by inducing the transformation of both epithelial and endothelial cells to myofibroblasts through epithelial–mesenchymal transition (EMT) and endothelial–mesenchymal transition (EndoMT) [24][25]. In extra-intestinal fibrosis, several studies highlighted the critical role of macrophages, especially M2 subsets, as a critical source of TGF-β1 [25][26]. In liver fibrosis, TGF-β1 has been described as having a dual antagonistic role. Indeed, TGF-β1-producing regulatory T (Treg) cells have been shown to decrease fibrosis through an interleukin (IL)-10-dependent mechanism, whereas macrophage-derived TGF-β1 exerts pro-fibrotic effects [26][27]. As a matter of fact, many studies have identified several macrophage subtypes as essential producers of TGF-β1 with the profibrotic cytokine IL-13, which is able to induce and activate latent TGF-β1 through an MMP9-dependent mechanism [27][28][28,29]. Altogether, this suggests that TGF-β1 activity inhibits inflammation if produced by Treg cells but promotes fibrosis when produced by macrophages. The fact that TGF-β1 can simultaneously suppress inflammation but promote collagen synthesis in myofibroblasts likely explains the dual role of this cytokine. Targeting the inhibition of the specific profibrotic macrophage, rather than globally alleviating TGF-β1, might provide a more rational approach to ameliorate fibrosis. Indeed, studies have already shown that decreasing the number of TGF-β1-producing macrophages significantly slows the progression of hepatic fibrosis [29][30]. Production of TGF-β1 is mainly conducted by M2 macrophages. During fibrogenesis of the heart and kidneys, disturbance to the balance M1/M2 macrophages have been observed, with a switch of M1 towards M2 [30][31][31,32]. In unilateral ureteral obstruction, an experimental model of renal fibrosis, higher levels of TGF-β1 were detected in M2 macrophages than in myofibroblasts [31][32]. In Hereinthis study, the invalidation of STAT6, a key transcription factor in M2 differentiation, decreased the production of collagen and the activation of TGF-β1/Smad signalling and inhibited renal fibrosis.
M2 macrophages can be further classified into four subtypes according to their patterns of biomarkers: M2a, M2b, M2c, and M2d. These macrophage subsets play distinct roles in the inflammatory response and wound-healing process (Figure 1). However, factors influencing subset polarisation, and their specific functions in vivo remain unclear. Nonetheless, in vitro, M2a macrophages are typically induced by IL-4 and IL-13; the M2b macrophages are induced in response to immune complexes and bacterial lipopolysaccharide (LPS), and the M2c macrophages to glucocorticoids, IL-10, and TGF-β. Lastly, the M2d macrophages are induced in response to IL-6 and adenosines [32][33]. Recent studies suggest that specific M2 subpopulations could be assigned to fibrogenesis progression while certain others are assigned to wound repair (Figure 1). In renal fibrosis, the roles of macrophages M2a and M2c have been recently described [33][34]. In a murine model of renal fibrosis, an increased M2a infiltration is observed and correlated with a higher fibrosis score [33][34]. In this Hereinstudy, the inhibition of M2a macrophage infiltration was inhibited following administration of trichostatin A, a histone deacetylase (HDAC) inhibitor. Accordingly, an upregulation of M2c macrophage populations in the kidneys was observed and was associated with a diminution of renal fibrosis [33][34]. Indeed, M2a macrophages produce pro-fibrotic factors, including TGF-β1, connective tissue growth factor (CTGF), fibroblast growth factor (FGF), and insulin-like growth factor (IGF) [34][35]. M2c macrophages act partly via STAT1 and NF-kB inhibition, leading to inactivation of myofibroblasts and restriction of M1 and M2a macrophage subsets via a local production of IL-10 [34][35]. In line with these observations, in cardiac fibrosis, the different M2 macrophage phenotypes have divergent effects on cardiac fibroblasts, especially the M2a (pro-fibrotic) and M2b (anti-fibrotic) phenotypes. Indeed, the intracardiac injection of M2b macrophages in a rat model of cardiac ischemia/reperfusion injury significantly suppressed the proliferation and migration of cardiac fibroblasts. Moreover, this approach attenuated the expression of fibrosis-related proteins and abrogated the differentiation of cardiac fibroblasts into myofibroblasts [35][36]. These studies suggest that M2a macrophages are profibrotic and could be critically involved in fibrogenesis. Thus, the dual role of M2 macrophages could be explained by the balance of distinct M2 subpopulations in chronic fibrotic disorders.
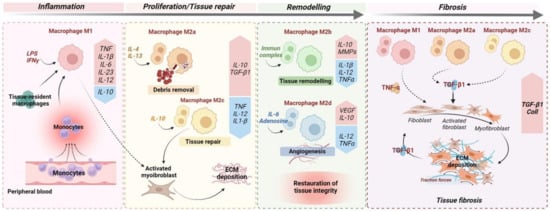
Figure 1. Monocyte and macrophage polarisation during wound healing and fibrosis. The optimal wound healing process could be dived into three distinct phases: inflammation, proliferation, and remodelling. After an acute injury (left), the first phase of wound healing is predominated by pro-inflammatory signals, including lipopolysaccharides (LPSs), that stimulate pro-inflammatory M1 macrophages but also the recruitment upon injury of circulating monocytes, which develop into pro-inflammatory M1 macrophages. In turn, M1 macrophages secrete high concentrations of pro-inflammatory cytokines, such as TNF and IL1-β. In the meantime, IL-4 and IL-13 induce M2a macrophages that clear apoptotic cells. During the proliferative phase, in response to anti-inflammatory cytokines/mediators, including IL-4 and IL-13, and the efferocytosis of apoptotic cells ensured by M2a macrophages, macrophages undergo functional reprogramming toward a pro-restorative phenotype: M2c. These, later, by secreting TGF-β1, promote activation of myofibroblasts and extracellular matrix (ECM) deposition. During the last phase of wound healing, IL-10 is a key anti-inflammatory cytokine produced during the proliferative stage of repair that facilitates tissue remodelling by activating M2b macrophages which release metalloprotease matrix proteins (MMPs) to regulate ECM degradation. In addition, angiogenic response is promoted by M2d macrophage-releasing pro-angiogenic factors, including VEGF. Ultimately, the remodelling phase concludes with complete restoration of tissue. When chronic injuries occur (right), the persistence of M1, M2a, and M2c macrophage activation leads to fibrogenesis through the secretion of pro-inflammatory (TNF) and pro-fibrotic (TGF-β1) cytokines, resulting in sustained myofibroblast activation and leading to excessive ECM deposition.
In intestinal fibrosis, the roles of M2 macrophages are not yet well elucidated. However, recent studies using an experimental rat model of IBD underlined the critical role of M2 macrophages in the progression of intestinal fibrosis and its associated complications, such as strictures. In the TNBS rat model, an important M2 phenotype of macrophages was specifically detected in the underlying smooth muscle at the stricture site [36][37]. HereIn thins study, the authors developed an in vitro model of the stricture phenotype using intestinal smooth muscle cells and showed that TGF-β1 secreted by M2 macrophages led to a hyperproliferative phenotype of these cells, whilst improving their survival in hypoxic conditions. In the dextran sulfate sodium (DSS)-induced colitis model, DSS administration directs the phenotype of macrophages towards the M2 lineage and seems to inhibit pathogen clearance and exacerbate inflammation, contributing to fibrosis development [37][38]. However, several other studies report the beneficial effects mediated by M2 macrophages. M2b macrophage-derived exosomes reduced the severity of DSS-induced colitis and promoted mucosal healing in mice [38][39]. These contradictory results may be explained by a disrupted balance of distinct M2 subpopulations in IBD. Thus, promoting the reprogramming of macrophages to an “anti-fibrotic M2 subset”, such as M2b, could be a potential strategy in the treatment of fibrogenesis, including intestinal fibrosis.
In intestinal biopsies from CD patients, M2 macrophages are found in fibrotic areas, but the causal effect of M2 macrophages and its subtypes in intestinal fibrosis remain unrevealed [39][40]. However, some lessons from extra-intestinal fibrosis, such as cardiac fibrosis, could be partially true for the gut. It is essential to emphasise that M2 macrophages assume a spectrum of phenotypes with different functions, whose equilibrium during the disease are key events for recovery. Thus, studies in which M2 macrophages are found to be beneficial for fibrosis may refer to an increase of antifibrotic activity and an alleviation of pro-fibrotic activity. That is why it appears essential to unravel the complexity behind the macrophage polarisation and most particularly the M2 subtypes which promote wound healing and counteract intestinal fibrogenesis.