Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Alejandro Martinez-Roca and Version 2 by Catherine Yang.
Lynch-like syndrome (LLS) is defined as colorectal cancer cases with microsatellite instability (MSI) and loss of expression of MLH1, MSH2, MSH6, or PMS2 by immunohistochemistry (IHC) in the absence of a germline mutation in these genes that cannot be explained by BRAF mutation or MLH1 hypermethylation.
- lynch syndrome
- colorectal cancer
1. Introduction
Lynch-like syndrome (LLS) is defined as colorectal cancer cases with microsatellite instability (MSI) and loss of expression of MLH1, MSH2, MSH6, or PMS2 by immunohistochemistry (IHC) in the absence of a germline mutation in these genes that cannot be explained by BRAF mutation or MLH1 hypermethylation [1]. Managing these cases is challenging because the subsequent carcinogenic process is yet to be unveiled. LLS is probably caused by somatic mutations in the mismatch repair (MMR) genes, and, therefore, it is sporadic [2][3][2,3]. However, patients with LLS and their relatives have an increased risk of colorectal cancer (CRC), suggesting a possibility of inherited risk. Thus, the most probable scenario is that LLS represents a mixture of sporadic MSI cases, unidentified Lynch syndrome (LS) cases, and possibly other hereditary cases of yet-to-be-determined origin [1][4][1,4]. Differentiating between both sporadic and hereditary origin has been a challenge, mainly due to the difficulty in conducting mutational somatic studies of CRC samples.
2. Lynch Syndrome
LS is the most common hereditary cancer syndrome and accounts for approximately 3% of all CRCs [5][22]. LS is an autosomal dominant disorder caused by germline mutations in MLH1, MSH2, MSH6, and PMS2, as well as deletions in EPCAM. Germline EPCAM deletions result in methylation of the surrounding genomic region, affecting the MSH2 promoter located 18 Kb downstream. As a consequence, MSH2 gene expression is silenced [6][23]. Constitutional epigenetic silencing of MLH1 [7][8][9][10][11][12][13][14][15][16][17][18][19][24,25,26,27,28,29,30,31,32,33,34,35,36] and hypermethylation of MSH2 as a consequence of EPCAM deletion [12][29] have been rarely reported in some families. LS patients develop multiple tumors, most frequently colorectal and endometrial [20][37], but also upper gastrointestinal, ovarian, biliary, urinary, brain, non-melanoma skin, and prostate tumors [21][38]. LS patients are diagnosed at an early age; with a mean age of diagnosis of around 45 years, they develop cancer a mean of 23 years earlier than the general population [22][39]. Lynch tumors develop faster than sporadic CRC [23][40]. LS patients have an increased risk of synchronous and metachronous neoplasias. Approximately 7% of LS patients have multiple cancers when diagnosed [23][24][40,41]. LS tumors are poorly differentiated, and some present with mucinous features, a medullary growth pattern or showing infiltrating lymphocytes [25][26][42,43]. Moreover, their location is predominately in the proximal colon [23][27][40,44]. The diagnostic algorithm for LS starts by testing tumors for MSI and/or loss of immunochemical expression of MMR proteins. The Jerusalem guidelines, so-called ‘universal screening’, recommend screening all CRCs and endometrial patients <70 years old for MSI or MMR-D [28][45]. If MLH1 is lost in IHC, the tumor should then be tested for methylation of the promoter of MLH1 and/or the BRAF V600E mutation to rule out sporadic CIMP tumors. If testing negative, patients are submitted to germline testing, which includes the sequencing and the analysis of deletions and duplications in the appropriate MMR genes. Germline testing results confirm an LS diagnosis [29][30][46,47], whereas, if MLH1 is methylated in a tumor, a complementary MLH1 methylation study in blood should be performed to identify constitutional epimutation of MLH1 [31][48] (Figure 12).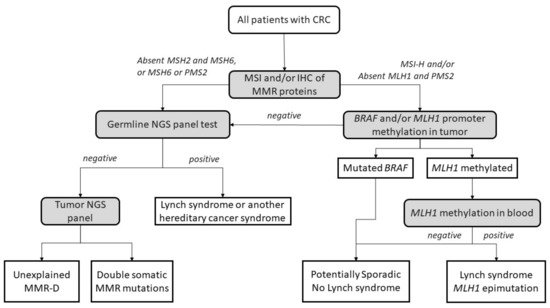
Figure 12. Universal screening strategy for Lynch-syndrome patients. CRC, colorectal cancer; MSI, microsatellite instability; IHC, immunohistochemistry; MMR, mismatch repair; MMR-D, mismatch repair deficiency; NGS, next-generation sequencing. Adapted from Valle et al. [32][49].
3. Potential Causes of Lynch-like Syndrome
Different plausible causes to explain the origin of LLS tumors have been described. According to the hereditary origin, unknown mechanisms or germline mutations in other genes than those involved in the classical MMR system could mimic the Lynch phenotype with MMR-D. In addition, some LLS cases could be LS with unidentified germline MMR mutations. In contrast, LLS could be due to somatic defects in genes related to tumor onset and progression or due to biallelic alterations in MMR genes outside MLH1 promoter methylation [2][3][33][2,3,58], thus having a sporadic origin. A frequent explanation for LLS cases that should always be ruled out is false-positive IHC/MSI results, which represent approximately 19% of cases in some series [34][59], and confirmation of MSI and IHC status should be the first step before classifying a case as LLS. Figure 2 3 describes different potential causes of LLS. It is important to clarify that, if some LLS cases after their molecular analysis can be classified in another category, they will no longer be considered LLS. They will be included in the surveillance program of the new group.
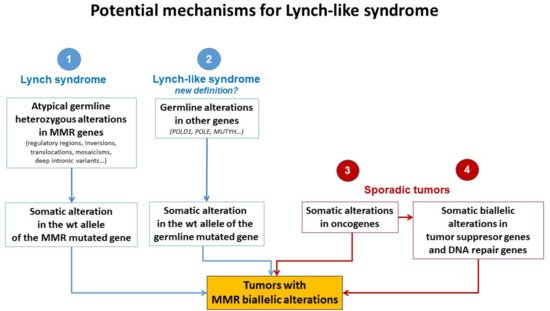
Figure 23. Potential mechanisms for Lynch-like syndrome. MMR, mismatch repair; wt, wild type. Adapted from Pico et al. [35].
Potential mechanisms for Lynch-like syndrome. MMR, mismatch repair; wt, wild type. Adapted from Pico et al. [54].
3.1. Germline Mutations in Other Genes Affecting the MMR System
The fact that LLS patients are younger at diagnosis than sporadic cases and some of them have a family history of LS-related neoplasias suggests that germline mutations in other genes could also be involved in cancer development in some of these cases (Figure 23). It is important to distinguish whether MMR-D is driving tumor formation or is a secondary event. Germline mutations in MUTYH and POLE have been reported in some patients with MMR-D [14][36][37][38][31,60,61,62]. Mutations in MUTYH have been previously associated with MUTYH-attenuated polyposis [14][38][31,62]. In addition, mutations in MUTYH have been described in MLH1-methylated tumors [14][38][31,62]. Approximately 1–3% of LLS cases carry biallelic mutations in MUTYH [14][38][31,62]. In addition, mutations in the exonuclease domain of POLE and POLD1 cause a hypermutator phenotype that confers a high predisposition to developing attenuated colorectal polyposis at an early age. POLE and POLD1 mutations may be associated with MMR-D in some cases due to MMR mutations secondary to the hypermutator phenotype [36][37][39][40][41][60,61,63,64,65].
On the other hand, Xavier et al., found potentially pathogenic variants in a group of genes involved in the regulation of cellular activity (EXO1, POLD1, RFC1, and RPA1) [42][55]. EXO1 is related to the union of MLH1 and MSH2, and a mutation in these genes may trigger MMR-D [43][66]. In addition, RPA1 and POLD1 are associated with harmful effects in tumors with mutations in these genes [44][45][67,68]. RFC1 has been described in the development of different malignancies. Huang et al., noted the presence of a variation of this gene in a plasmatic cell tumor [46][69]. Moreover, somatic mutations in RFC1 were reported in 10.2% of uterine carcinomas and 5.5% of CRCs [47][70]. This gene also plays an important role in genomic integrity because it is a member of the BRCA1-associated genomic surveillance complex [48][71]. Golubicki et al., found unknown variants in four genes (POLE, ERCC6, RAD54L, and PALB2) in a group of LLS patients [49][72]. ERCC6 and PALB2 have been associated with CRC [37][50][51][61,73,74], and the PALB2 variant was previously reported in a suspected case of LS [52][75].
Next-generation sequencing (NGS) studies have allowed the identification of pathogenic variants that could be candidates for familial CRC with unknown genetic basis. Recently published studies have identified pathogenic variants in genes that maintain DNA integrity resulting in a variety of clinical phenotypes. Germline variants in NTHL1 cause adenomatous polyposis and CRC [53][76]; MCM9 variants are associated with hereditary mixed polyposis, CRC, and primary ovarian failure [41][54][65,77]; and variants in FAN1 cause hereditary CRC by impairing DNA repair [55][78]. Following this line of inquiry, variants in BUB1 and BUB3 [56][79], SETD2 [57][80], WRN [58][81], BARD1 [58][81], MCPH1 [58][81], and REV3L [58][81] have been found in the germline analysis of LLS cases, linking the mutation of WRN, BARD1, MCPH1, and REV3L for the first time with CRC.
3.2. Hereditary Cases: Unknown Mutations in MMR Genes
In some cases, LLS patients are actually LS patients whose pathogenic variants have not been identified (Figure 23). Current techniques cannot easily identify complex and cryptic mutations. Intronic regions, structural changes such as inversions, and/or copy number variation (CNV) are rarely analyzed genetic changes but may play an important role in unveiling mutations in these patients. For example, the mutation 478 bp upstream of exon 2 in MSH2 creates a canonical splice donor site. The pseudo-exon that is created contains a stop codon that results in a truncated protein [41][59][65,82].
Structural changes have been found in some families, such as the inversion of MSH2 exons 1–7 in 10 families in North America [60][61][62][83,84,85] and the inversion of MSH2 exons 2–6 in two families in Australia [63][86]. Another example of structural genetic changes is the MLH1-LRRFIP2 fusion after a paracentric inversion of chromosome 3 [64][87] or deletion in that same locus [41][65][65,88]. Moreover, Hellen et al., show a retrotranspositional insertion in PMS2 mediated by LINE-1 between exon 7 and 8 [66][89].
Regulatory regions of MMR genes should also be taken into account. In some cases, variants in the promoter region of MMR have been associated with reduced promoter activity or transcriptional silencing of the allele [57][67][80,90]. The accumulation of mutations in the 3′UTR of genes affects mRNA stability and, therefore, protein expression. Germline 3′ UTR mutations in MLH1 have been associated with loss of expression [68][91]. On the other hand, abnormal regulation of protein expression by miRNA could cause a loss of MMR gene expression. High levels of miRNA-21 downregulate MSH2 and MSH6 and have been found in CRC with loss of MSH2 expression [69][92]. The same has been seen with MLH1 and miRNA-155 [69][92]. These examples show the importance of more extensive sequencing methods to detect complex mutations in families of patients with MMR-D and without germline mutations by routine procedures.
Somatic mosaicism could also account for some LLS cases. For instance, Sourrouille et al., described a case of somatic mosaicism in MSH2 after de novo mutation of this gene [33][58]. Another study described somatic mosaicism in a woman with synchronous gynecological tumors at 44 years old. The MLH1 mutation was only present in 20% of the allele fraction in normal tissue, but her sister and father, who were also affected with LS-related tumors, carried the same mutation [70][93]. A recent study reported a case of de novo somatic mosaicism in which the MLH1 mutation was detected in the tumor and at a lower level in peripheral blood but not in any other family member [71][94]. Mosaicism can be detected using highly sensitive NGS with high coverage, and more genetic-driven cases could be correctly identified.
Another important factor to consider is the presence of variants of uncertain significance (VUS) in approximately 30% of cases [72][95]. Some of them could be pathogenic but cannot be classified due to the absence of clinical, molecular, or functional evidence. Families carrying VUS are managed based on their family history of cancer until further variant classification is available [41][65].
3.3. Somatic Alteration in Other Cancer Genes or Epigenetic Structures
AT-rich interaction domain 1A (ARID1A) is mutated in a large proportion of tumors [73][100]. These proteins interact with MSH2, recruiting it to chromatin during DNA replication. Shen et al., demonstrated that impairment of ARID1A contributes to MMR-D [74][101]. In addition, a somatic exonuclease domain mutation in POLE would be involved in phenocopying defective MMR DNA in 25% of unexplained endometrial cancers with MSI [75][102].
Local inflammation also promotes genetic and epigenetic alterations in CRC [76][103] and has been determined to be an important factor in damage to the MMR system [77][104]. An increase in the concentration of proinflammatory cytokine IL-6 has been demonstrated to alter MMR function. IL6 can activate STAT3, which drives MSH3 out of the nucleus and prevents it from performing its nuclear function [78][105]. In the same way, high levels of reactive oxygen species (ROS) can induce DNA damage, resulting in MMR-D. Chang et al., showed that non-cytotoxic H2O2 can damage MMR complexes, triggering a reduction in these proteins [79][106].
Somatic methylation could also explain the MMR-D present in some LLS cases (Figure 23). Many tumor suppressor genes are methylated in sporadic cancers, including RB [80][81][107,108], VHL [82][109], and BRCA1 [83][110], as well as MLH1 promoter hypermethylation in sporadic CRC caused by the CIMP phenotype [84][111]. Recently, Buckley et al., reported an association between the methylation of SHPRH and MSI burden [85][112]. In addition, epimutations in MLH1 and MSH2 have been reported in some families [7][8][9][10][11][12][13][15][16][17][18][19][24,25,26,27,28,29,30,32,33,34,35,36], but other MMR genes can also be targets of somatic methylation [41][65].
3.4. Somatic Biallelic Alteration in MMR
When comparing tumors with double somatic alterations to LS tumors, no significant histopathological difference was found [86][116]. Tumor sequencing is the adequate way to evaluate double somatic mutations. Therefore, tumor sequencing should be considered to clarify sporadic versus hereditary causes of unexplained MMR-D [41][87][65,117].
Different studies investigated promoter methylation of MMR genes in LLS patients. Methylation of MSH2 was only found in one [87][88][117,118] out of 53 LLS patients with loss of expression of MSH2 by IHC studied [57][87][88][80,117,118]. The MSH6 promoter was unmethylated in 108 patients with LLS and MMR-D [57][89][90][80,119,120], and the same happened with the PMS2 promoter in 100 cases with loss of expression of PMS2 or MLH1 who were negative for PMS2 promoter methylation [91][121]. In summary, based on these studies, somatic promoter hypermethylation of MMR genes does not seem to be the underlying cause of MMR-D in these unexplained tumors.
Therefore, there is a subgroup of LLS that can be explained by double somatic inactivation, and these cases should probably be excluded from the LLS classification due to the probable sporadic origin. However, this approach still has some open questions, because there is no standardized universally accepted technique or protocol for differentiating these cases. Moreover, the biallelic somatic inactivation of MMR genes can also be due to any of the previously described mechanisms, some of them generated by germline genetic alterations. Classifying patients as sporadic or potentially hereditary cases should also be the subject of clinical validation by adequately comparing pedigrees, with long-term follow-up of these families in order to find differences in the incidence of CRC and other LS-related disorders. When some groups advocate for generalization of a somatic study of LLS cases, it is necessary to reach a consensus on how to perform such a study and which cases could be confidently considered as sporadic with no indication for follow-up of patients and relatives. This algorithm has not been clinically validated. Table 1 show a summary of potential causes of LLS.
Table 1.
Potential causes of LLS. LLS, Lynch-like syndrome; MMR, mismatch repair.
| Mutations in other Genes Affecting MMR System (Germline) |
Unknown Mutations in MMR Genes (Germline) |
Somatic Mutations in Cancer Genes (Somatic) |
Biallelic Alteration in MMR (Somatic) |
|
|---|---|---|---|---|
| MUTYH | Mutation EXON 2 MSH2 | H3K36me3 | Double somatic hit | |
| POLE/POLD1 | Inversion EXON 1-7 MSH2 | SETD2 | Somatic mutations in MMR genes | |
| EXO1/RFC1/RPA1 | Inversion EXON 2-6 MSH2 | PCNA | Methylation in MMR genes | |
| ERCC6/RAD54L/PALB2 | MLH1 | -LRRFIP2 fusion | ARID1A | |
| PIK3CA | MLH1 | 3′ UTR mutation | POLE | |
| FAN1/MCM9 | Deep intronic variant in MSH2 | IL-6 and oxidative stress | ||
| NTHL1 | miRNA 21 AND miRNA 155 | Methylation in other genes | ||
| BUB1/BUB3/WRN/MCPH1/REV3L | Mosaicism | |||
| VUS |