Large Conductance Calcium and Voltage-Gated Potassium Channels (BKCa). Also known as Slowpoke, KCa1.1, and MaxiK channels.
- BK Channels, Cardio protection, ROS, Modulators
- BK Channels
- Cardio protection
- ROS
- Modulators
1. Introduction
In the early 1980s, ‘big K’ channel (BKCa), named after its large single-channel conductance 250–300 pS (in symmetrical 150 mM KCl), was originally cloned in Drosophila at the slowpoke (slo) [1,2]. BKCa channels are ubiquitously expressed in a broad range of excitable and inexcitable cell types [4], as well as in intracellular organelles, including mitochondria [5], nuclei [6], endoplasmic reticulum [7,8], and lysosomes [2,9,10], where they are termed as iBKCa [10]. BKCa channels participate in a wide variety of fundamental physiological processes from vascular tone [11,12], cardiac rhythmicity [11,13], erectile and urinary autonomic functions [14,15], regulation of gene expression [6], and aging [16]. Recent reports suggest that alterations of BKCa function and expression may associate with several pathological conditions, such as paroxysmal nonkinesigenic dyskinesia with a gain of function, and cerebellar ataxia with loss-of-function mutations [17,18], making them unique therapeutic targets.
2. Role of BKCa Channel Agonists
The ubiquitous BKCa expression, a dual activation mechanism that allows them to couple intracellular signaling to membrane potential and significantly modulate physiological responses in a plethora of tissues, prompted intense development of BKCa channel modulators [73,74]. The number of identified molecules is significant and growing, which is why we will limit this review to regularly used pharmacological tools (Figure 21) to study BKCa.
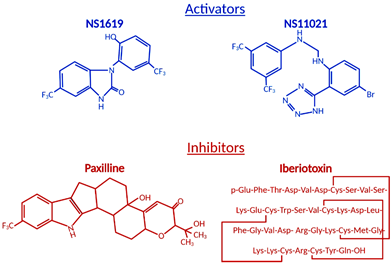
Figure 21.
Structural formulas of commonly used mitochondrial BK
Ca
modulators.
Among the first synthetic BKCa activators were benzimidazoles NS004 [75] and NS1619 [76] (Figure 21) and the latter became one of the most used agonists in establishing the physiological functions of BKCa. NS1619 accelerated K+ mitochondrial uptake, improving mitochondrial respiratory function, but its link to cardioprotection was made when NS1619 was administered prior to the ischemic event and protected isolated perfused rat hearts from global I/R injury [38]. To further elucidate cardioprotective mechanisms, several studies probed ROS production and mitochondrial Ca2+ levels during ischemia and reperfusion. Isolated guinea pig hearts were subjected to I/R injury in the presence and absence of NS1619 [41]. Hearts were nearly continuously monitored for levels of nicotinamide adenine dinucleotide (NAD) + hydrogen (H) (NADH), superoxide, and mitochondrial calcium and NS1619 showed attenuated levels when compared with untreated hearts that resulted in an astounding 50% decrease in infarct size [41]. Those effects were nullified by paxilline and superoxide dismutase, showing that both BKCa channel activity and superoxide are necessary for the cardioprotective effect [41].
In addition to previously mentioned synthetic activators, a plethora of endogenous modulators have been studied that induce similar changes in BKCa. These molecules include gasotransmitters, such as nitric oxide (NO), carbon monoxide (CO), and hydrogen sulfide (H2S) [90]. CO has been shown to increase the open probability (PO) of BKCa by mimicking the action of Ca2+ because the mutation of residues responsible for Ca2+ sensitivity also prevented channel CO sensitivity [91,92]. Furthermore, the same mechanism appears to be responsible for BKCa activation by H+, which could contribute to BKCa’s cardioprotective function, as intracellular pH falls by 0.5 to 1 unit during the early stages of ischemia and changes in extracellular acidosis had no significant effect on BKCa [92–96]. NO is a well-established vasodilatory factor released by endothelial cells and it has been shown to significantly increase PO of BKCa by direct interactions and/or through cyclic guanosine monophosphate (cGMP)-mediated phosphorylation [97,98]. Another molecule from the gasotransmitter group is H2S, a biologically active gas that plays a role in the physiology and pathophysiology of cardiovascular and nervous systems [99,100]. There are conflicting data showing contradictory results that vary from inhibition of native and recombinant currents of the α subunit expressed in human embryonic kidney (HEK)-293 cells [101], through to β1 subunit presence and enhanced oxidative regulation and BKCa activation [102], to the BKCa response dependent on channel priming by PKG phosphorylation [103]. These findings indicate the complexity of BKCa signaling, which heavily depends on allosteric interactions, oxidative state, phosphorylation, divalent ion concentration, and voltage. MitoBKCa, because of their localization, are continuously exposed to ROS.
3. Role of BKCa Channel Antagonists
Antagonists and inhibitors (Figure 21) of BKCa channels are widely used for cardioprotection studies. One of the most widely used small molecules and synthetically derived BKCa channel blockers is tetraethylammonium chloride (TEA) [106,107]. TEA blocks channel activity by binding within the ion conduction pathway in a voltage-dependent manner from both sides of the membrane; however, it lacks selectivity for BKCa channels as TEA blocks several voltage-gated potassium channels [108,109]. Venom from scorpions is an invaluable source of BKCa inhibitors. The first potent BKCa blocker was a 37-amino-acid peptide charybdotoxin (ChTX) identified in 1985 [110]; however, because of its inhibitory effect on Kv1.3 and intermediate-conductance Ca2+-activated-K+ channels (IK channels), currently, ChTX use in BKCa-specific studies is limited [111]. Another 37-aa peptide isolated from scorpion venom was iberiotoxin (IbTX), which showed high selectivity and affinity for BKCa channels [112]. The blocking mechanism of ChTX and IbTX is attributed to their binding to the external pore region of the channel through bimolecular reaction and physically blocking the conduction pathway [113]. Toxins, because of their peptide properties, such as rapid degradation, poor reversibility, and blood-brain barrier, are less than ideal for extended research use or drug development.
The next group of non-peptide BKCa channel inhibitors is a family of tremorgenic mycotoxins isolated from fungi and this group includes potent neurotoxin paxilline (PAX) [114]. Paxilline is the non-peptide neurotoxin most extensively used in research because of its high selectivity and reversibility of action, and capability of a 70% BKCa channel current inhibition at a concentration as low as 10 nM (Ki = 1.9 nM), and its site of action is located on the α-subunit and cytoplasmic side [115,116]. IbTX and PAX were used in multiple studies to determine the role of BKCa channels in the cardiovascular system. PAX resulted in a significant decrease (30%) in the heart rate of wild-type mice with no effect on mean blood pressure [57]. This effect was transient and concentration-dependent. To remove the possibility of systematic effects of PAX, isolated and perfused rat hearts also showed a decreased heart rate due to PAX (34%) and IbTX (60%) treatment [57]. IbTX is not cell permeable and those results suggested that BKCa channels expressed on the cell membrane of SAN cells play a role in heart rate regulation, not channels presented on IMM as we have seen in adult cardiomyocytes. Those findings were reproduced with a similar heart rate reduction caused by PAX, further supporting the presence of BKCa on the plasma membrane of SAN cells [11].
BKCa blockers were used in combination with activators to farther validate BKCa channels’ involvement in the area of interest that especially applies to I/R injury research. As we previously mentioned, BKCa was localized in IMM of cardiomyocytes, and treatment with NS1619 (BKCa opener) protected the heart during I/R injury, reflected in the significantly reduced infarct size [38]. This effect was reversed by PAX, which allowed the identification of BKCa channels as a major player in cardioprotection, and since then, those finding have been confirmed in numerous animal models [38,41,43,77–81,122,123]. For clarification of the mechanism by which BKCa channels resulted in a cardioprotective effect, close monitoring of mitochondrial changes during I/R injury was required. Continuous monitoring of NADH, superoxide, and mitochondrial Ca2+ levels in guinea pig hearts subjected to I/R injury revealed a significant reduction in mitochondrial Ca2+ capacity, ROS production, and levels of NADH on treatment with NS1619, which affected the end result: Infarct size was reduced by more than 50% in comparison to the control group [41]. Those cardioprotective effects were negated by PAX and superoxide dismutase, which indicated that both BKCa channels and superoxide activity are necessary to elicit cytoprotective effects [41].
4. BKCa as a Therapeutic Target for Cardioprotection
The first preconditioning intervention was reported in 1986 when it was demonstrated that four short ischemic-reperfusion pulses resulted in a dramatic 75% decrease in infarct size [130]. The nature of this protective mechanism suggested the involvement of the K+ channel. The role of BKCa channels in cardiomyocytes was neglected because of their absence from the plasma membrane of cardiomyocytes [10]. However, in 2002, O’Rourke’s group reported voltage- and calcium-dependent potassium currents on mitoplasts isolated from guinea pig cardiomyocytes [38]. The recorded currents had a single-channel conductance of ~307 pS and were blocked by ChTX [38]. Furthermore, NS1619 protected the heart from the ischemic event and this effect was blocked by PAX [38]. Subsequent studies confirmed that NS1619 protected the heart from I/R injury in a number of animal species and administration of NS11021, before or right after, also protected the heart from I/R injury [50,131]. Because most of the data addressing the role of mitoBKCa channels in cardioprotection relied on pharmacological tools, the data published were questioned because both activators and blockers displayed unspecific effects. Our group resolved controversies about pharmacology, showing that the cardioprotection offered by NS1619 was lost in Kcnma1-/- mice and clearly demonstrating BKCa channels’ involvement in cardioprotection, which was also confirmed with the use of cardiomyocyte-specific knockouts, CM-Kcnma1-Crefl/fl [5,47]. Additionally, changes in ROS production and attenuated oxidative phosphorylation in Kcnma1-/- cardiomyocytes were also observed, suggesting the role of mitoBKCa in fine-tuning the oxidative state of the cell [42]. We still do not have a clear picture of how the BKCa channel contributes to cardioprotection, although a few mechanisms have been proposed. The BKCa channel affects mitochondrial Ca2+ accumulation, K+ influx leads to partial depolarization if the IMM reduces the driving force for Ca2+ entry, and prevents Ca2+ overload during I/R injury [41,44]. Additionally, pre-treatment with NS1619 increased mitochondria Ca2+ retention capacity, an effect that was lost in Kcnma1-/- mice [5].
The role of mitoBKCa has also been shown in the regulation of ROS production in various models. Isolated mitochondria from cardiomyocytes and neurons showed decreased ROS production when stimulated with NS1619, which was confirmed in isolated hearts and a Kcnma1-/- mice model, where mitochondria isolated from knockout mice showed elevated ROS production after the anoxic event [41,42,131]. Moreover, mitoBKCa opening has been shown to improve mitochondrial energy production by swelling of the mitochondrial matrix [61]. An attenuated phosphorylation capacity was also identified in a CM-Kcnma1-Crefl/fl mice model, which further connected mitoBKCa activity with ATP preservation [42]. During reperfusion, after an ischemic event, mitoBKCa activity would improve oxidative phosphorylation, decrease ROS production, and improve mitochondria Ca2+ retention, preventing opening of the mitochondrial permeability transition pore (mPTP), which would lead to mitochondria collapse, termination of ATP synthesis, and cardiomyocyte death [40]. The recent findings with the use of pharmacological tools and genetic models have clearly demonstrated that BKCa channels are important for cardioprotection, regulation of vascular tone, and mitochondrial function. However, there are still variations in results that seem to depend on the methods chosen and experimental design, and imperfect activators and blockers that display a number of unspecific side effects. The number of synthetic BKCa activators have been developed through the years by different pharmaceutical companies, with promising results in animal models, but with limited success in clinical trials [131]. The research established mitoBKCa channels as a promising target for limiting reperfusion damage and correcting long-term events that occur after AMI, but further research will be needed to develop clinical pharmacological tools for cardiac disease in the future.