Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Jessie Wu and Version 3 by Jessie Wu.
The Golgi apparatus is a central hub for cellular protein trafficking and signaling. Golgi structure and function is tightly coupled and undergoes dynamic changes in health and disease. A crucial requirement for maintaining Golgi homeostasis is the ability of the Golgi to target aberrant, misfolded, or otherwise unwanted proteins to degradation. Recent studies have revealed that the Golgi apparatus may degrade such proteins through autophagy, retrograde trafficking to the ER for ER-associated degradation (ERAD), and locally, through Golgi apparatus-related degradation (GARD).
- Golgi
- proteasomal degradation
- EGAD
1. Introduction
The Golgi, first described in 1898 by Camillo Golgi, is a stacked membranous organelle that serves as a hub of protein trafficking and post-translational modifications [1][2][1,2]. While traversing the Golgi, secretory glycoproteins undergo a series of modifications, wherein sugars are removed and added to their glycan chains, resulting in microheterogeneity of secreted glycoproteins [3]. Secretory proteins are sorted at the Golgi and are targeted to their final destinations, which include the plasma membrane, the endomembrane system, and secretion to the extracellular milieu.
Secretory proteins undergo strict quality and quantity control processes that monitor their proper folding, complex assembly, post-translational modifications, and correct targeting to organelles [4][5][6][7][4,5,6,7]. The first quality control checkpoint that secretory proteins undergo occurs co-translationally, during their synthesis by endoplasmic reticulum (ER)-bound ribosomes. Properly folded proteins exit the ER via ER exit sites (ERES) towards the Golgi apparatus. Damaged or unfolded proteins, however, are targeted by quality control machinery to degradation by the two main protein degradation pathways, namely the ubiquitin proteasome system (UPS) and lysosomal degradation. Canonically, cellular proteasomes are considered the main degradation machinery for cytosolic proteins, while transmembrane or secreted proteins are thought to be targeted to lysosomal degradation. However, this distinction is more complex when considering proteins along the secretory pathway, wherein proteasomes facilitate a large part of ER-associated degradation (ERAD). During ERAD, proteins that fail to pass the quality control checkpoint at the ER are translocated across the ER membrane and are degraded by proteasomes in proximity of the ER (Reviewed in [8][9][8,9]). Yet, some proteins are degraded by alternative lysosomal degradation routes. Lysosomal degradation of secretory proteins may be mediated either by direct vesicular trafficking to lysosomes or via autophagy, a process in which double membrane vesicles are formed de novo around substrates that are recognized by the autophagic receptor protein p62, or even around parts of organelles such as the ER. Newly formed autophagosomes then fuse with cellular lysosomes leading to the degradation of the engulfed proteins/content by lysosomal proteases [10][11][12][10,11,12].
2. Proteasomal Degradation and the Golgi
ERAD is a major mechanism for quality and quantity control of proteins in the secretory pathway. ERAD facilitates the degradation of ER membrane or luminal proteins via proteasomes, which are recruited to the ER membranes [13]. Over the years, numerous lines of research have alluded to the question of what happens to damaged proteins that escape the ER, or how is quality control of post-ER processes that occur at the Golgi, such as different modifications, regulated. Many studies describe that while the Golgi can serve as a sensor of quality control, proteasome-dependent degradation requires an additional step of retrieval of proteins from the Golgi to the ER, and their subsequent degradation by proteasome-dependent ERAD. For example, studies in yeast demonstrated that ERAD degradation of proteins that are expressed in excess, such as mutant vacuolar carboxypeptidase Y (CPY*) and Proteinase A (PrA*), requires cycling between the ER and Golgi [14][15][16][17][14,15,16,17]. Interestingly and perhaps counter-intuitively, for some proteins, exit from the ER was proven as a pre-requisite for degradation by ERAD [18][19][18,19]. For example, unassembled MHC molecules are degraded by ERAD only after reaching the cis-Golgi and subsequent retrieval to the ER [20]. Such findings raise the notion that there is more than merely overflow of the ER in the ‘decision’ to exit the ER prior to proteasomal degradation. It is yet unclear why some proteins are targeted to ERAD in the ER, while others must first reach the Golgi. IgM degradation in B cells is an interesting example that demonstrates the complexity of degradation routes in the secretory system. Specifically, the route of IgM degradation in B cells is associated with the differentiation stage of the cells. In early differentiation stages, pre-B cells produce an excess of a soluble form of IgM, which is efficiently degraded [21]. The degradation of the soluble IgM μ heavy chain was shown to be proteasome-dependent and to occur at a post-ER, pre-trans-Golgi, compartment [21][22][23][21,22,23]. Here, too, as in the case of MHC, it appears that proteins are retrieved to the ER before reaching the trans-Golgi, suggesting a role for the cis-Golgi in sorting substrates for ERAD. In support, retrieval to the ER and ERAD of misfolded transmembrane proteins occurs through their recognition by either the K/HDEL retrieval receptor, Rer1 [24], or Erv29, a COPII vesicle cargo receptor [17][25][17,25], both residing at the cis-Golgi.
The main signal associated with targeting of proteins to proteasomal degradation is ubiquitination by E3 ubiquitin ligases. Various ubiquitin E3 ligases, as well as deubiquitinating enzymes (DUBs) are known to be recruited to the Golgi apparatus and regulate the degradation of proteins. Yet, for many years, Golgi-localized ubiquitination was mainly reported in the context of trafficking or lysosomal degradation [26][27][26,27]. Nevertheless, several studies described phenomena in which proteins at the Golgi were targeted for proteasomal degradation that did not involve the ER or lysosomes. For example, in fission yeast, the sterol regulatory element-binding protein (SREBP) is proteolytically cleaved by Rhomboid 2 and Cdc48, following ubiquitination by the E3 ligase Dsc, which is localized to the Golgi apparatus [28][29][28,29]. The C-terminal fragment of SREBP is transported back to the ER following Rhomboid cleavage and is degraded by ERAD [30] while the N-terminus of the cleaved SREBP is translocated to the nucleus to act as a transcription factor [28]. Interestingly, in the absence of Rhomboid cleavage, the SREBP precursor is targeted to proteasomal degradation in a manner that is dependent on Dsc E3 ligase activity, and independent of ERAD E3 ligases Hrd1 and Doa10 [28]. The route that the SREBP precursor undertakes from the Golgi membrane to the proteasome was, however, not described.
The identification of endosome and Golgi-associated degradation (EGAD) in budding yeast [31] sheds new light on the role of proteasomes in Golgi-associated degradation (Figure 1). In EGAD, Golgi membrane proteins undergo proteasomal degradation without retrieval to the ER, but rather through their targeting from the Golgi to cytosolic proteasomes [31]. Interestingly, the first demonstration of EGAD involved the E3 ligase Dsc, which, as mentioned above, was likewise reported in fission yeast to mediate the ERAD-independent proteasomal degradation of SREBP by an uncharacterized mechanism [28]. While SREBP homologs are absent from budding yeast, the Golgi-localized Dsc E3 ligase complex was demonstrated in this strain to induce the polyubiquitination of the Golgi membrane protein Orm2 [31]. Yet, in contrast to most cases wherein polyubiquitinated proteins at the Golgi are sorted by ESCRT components to vacuolar/lysosomal degradation, Orm2 is extracted from the membrane following its ubiquitination via the function of the ATPase VCP/CDC48 and is targeted for degradation by cytosolic proteasomes [31]. As Orm2 is a negative regulator of sphingolipid biosynthesis, the post-ER checkpoint of EGAD is key to regulate lipid metabolism in budding yeast. It is plausible that Orm2 degradation may be the consequence of sphingolipid-sensing quality control mechanisms at the Golgi. Additional EGAD substrates and a potentially homologous mechanism in mammalian cells remain to be fully elucidated.
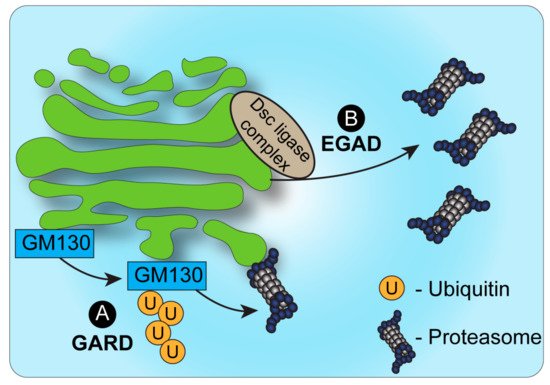
Figure 1. Proteasomal degradation and the Golgi. (A). Under conditions of Golgi stress, the structural Golgin protein GM130 is ubiquitinated and targeted for degradation by proteasomes, bound to the cytosolic side of the Golgi membrane. This process, known as Golgi apparatus-related degradation (GARD) allows the Golgi to regulate its morphology quickly in response to stress [32]. (B). In yeast, endosome and Golgi-associated degradation (EGAD) has been described as a mechanism by which proteins can be ubiquitinated by the Dsc complex, released from the Golgi membrane by VCP/CDC48 and degraded by cytosolic proteasomes [31].
The identification of EGAD demonstrated that proteins may be targeted to proteasomal degradation independently of ERAD. Yet, in contrast to ERAD, in the case of EGAD, proteasomes were not shown to be associated with the Golgi or endosomes, but were mainly localized to vesicles in the cytosol. A possible explanation may be the fact that the Golgi apparatus in yeast has a simplified, unstacked, architecture and is localized randomly in proximity to the ER. How then is ERAD-independent proteasomal degradation of Golgi-localized proteins regulated in mammalian cells? Recent findings demonstrated a novel mechanism of Golgi apparatus-related degradation (GARD) (Figure 1) that involves proteasomal degradation via proteasomes that are associated with Golgi membranes [32]. Specifically, proteasomes at the Golgi compartment were shown to be activated in response to Golgi-stress stimuli, such as block of Golgi trafficking or inhibition of sialylation, leading to the activation of GARD [32]. Such stress-induced activation of GARD was shown to be critical for regulating Golgi morphology, which is maintained by a series of structural proteins such as GM130 [32][33][32,33]. Golgi-stress induced the Golgi-localized degradation of the Golgi tethering factor GM130 through GARD, leading to Golgi dispersal. The block of proteasome activity, on the other hand, prevented Golgi dispersal under Golgi stress. Interestingly, these findings support and perhaps relate to previous work in neurons that described the proteasomal degradation of another Golgi tethering factor, GRASP65, following its ubiquitination by the Cul7-FbxW8 E3 ligase complex [34]. In that case too, the regulation of GRASP65 turnover controlled Golgi morphology [34]. Whether GRASP65 is targeted to degradation via Golgi-localized proteasomes in a Cul7-FbxW8-dependent manner remains to be determined.
Interestingly, Golgi dispersal via GARD is reversible, allowing the Golgi ribbon to recover its distinctive morphology when the stress is removed. In contrast, extended stress leads to irreversible changes and induces cell death in a manner that is dependent on GM130 degradation. Thus, localized proteasomal degradation allows for a rapid response to Golgi stress, providing a temporal window of response to control cell fate by either regaining Golgi integrity or inducing cell death. GARD-dependent regulation of Golgi stress was shown to be key in the highly secretory malignancy, multiple myeloma. Activation of GARD under Golgi-stress conditions led to cell death in multiple myeloma cells [32]. It is plausible to assume that multiple myeloma cells are particularly sensitive to changes in the Golgi, such as those induced by GARD, due to their extended secretory system, and their high dependence on intact Golgi structure and function. The Golgi stress-induced mortality of multiple myeloma cells proved beneficial in a mouse model of multiple myeloma, wherein treatment with the Golgi-stress inducer monensin dramatically reduced the number of malignant cells in the mouse [32]. While this work demonstrated the role of GARD in controlling cell death via regulation of Golgi morphology, the full range of GARD substrates and the effect of their degradation remains to be elucidated. Further, whether GARD serves to control only Golgi morphology, or may also provide a mechanism of quality control for proteins traversing through the Golgi remains to be investigated.
3. Quality Control and Stress Response at the Golgi
Protein quality control (PQC) mechanisms exist in numerous cellular compartments, beyond the ER. Ribosomes, mitochondria, the plasma membrane [5][35][36][37][5,35,36,37], and the Golgi [4] have all been reported to be involved in cellular homeostasis through localized PQC mechanisms. Such mechanisms provide continuous control over the fidelity of protein function, downstream of ERAD. For example, in yeast, the expression of a mutated, unstable form of bovine pancreatic trypsin inhibitor (BPTI) protein caused the accumulation of this secretory protein in the Golgi, from which mutant BPTI was targeted for vacuolar degradation [38]. Another secretory protein in yeast, Wsc1p, is able to evade ER quality control mechanisms despite mutations that cause misfolding of its luminal domain. Mutated Wsc1p is targeted to vacuolar degradation via the Golgi, suggesting that this protein undergoes its major quality control process in the Golgi, as opposed to the ER [39]. In mammalian cells, Briant et al. introduced different transmembrane domains into the secretory co-receptor CD8. While two of these domains caused retrieval of CD8 from the Golgi to the ER, a third domain targeted CD8 to lysosomal degradation directly from the Golgi [40]. These findings indicate that the Golgi quality control machinery can differentially target proteins to retrograde trafficking vs. degradation based on their transmembrane domains. In a recent study, Hellerschmied et al. used the Golgi-targeting sequences of MAN2A1 and either B4GALT1 or ST6GAL1, to target EGFP proteins to either cis- or trans-Golgi, respectively. These proteins also expressed a HaloTag2 domain, which can be chemically induced to unfold and expose hydrophobic domains [41]. Using this system, the authors could target Golgi-localized EGFP to lysosomal degradation and showed that unfolded proteins are identified and segregated from folded proteins within the Golgi, a critical ability in protein quality control, reminiscent of the ability of the ER to segregate and compartmentalize ERAD substrates [42].