Bacteriophages, abbreviated as “phages”, have been developed as emerging nanoprobes for the detection of a wide variety of biological species, such as biomarker molecules and pathogens. Nanosized phages can display a certain length of exogenous peptides of arbitrary sequence or single-chain variable fragments (scFv) of antibodies that specifically bind to the targets of interest, such as animal cells, bacteria, viruses, and protein molecules. Metal nanoparticles generally have unique plasmon resonance effects. Metal nanoparticles such as gold, silver, and magnetism are widely used in the field of visual detection. A phage can be assembled with metal nanoparticles to form an organic–inorganic hybrid probe due to its nanometer-scale size and excellent modifiability. Due to the unique plasmon resonance effect of this composite probe, this technology can be used to visually detect objects of interest under a dark-field microscope.
- bacteriophage (phage)
- biological detection
- biosensor
- nanomaterials
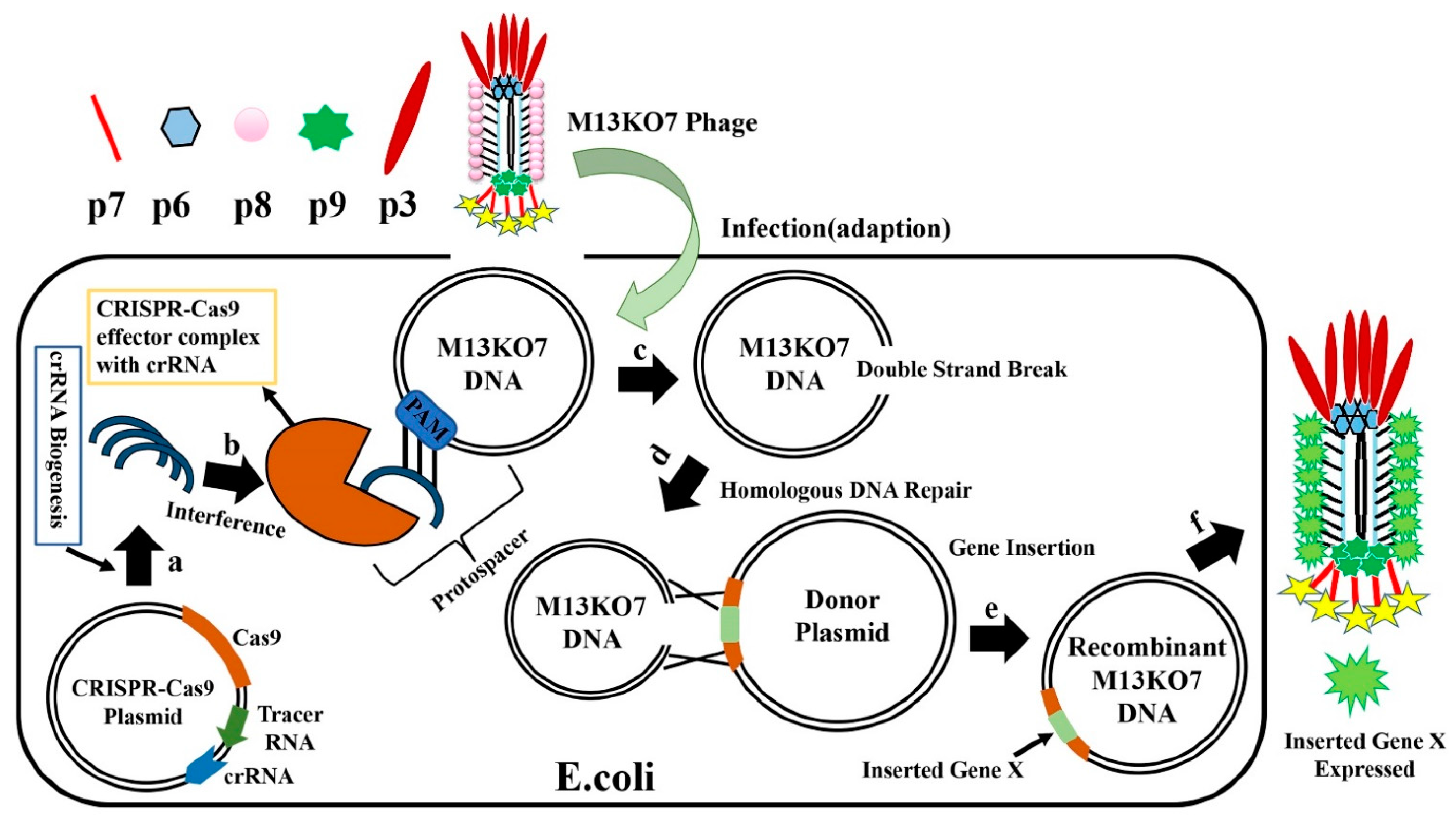
1. Homologous Recombination Technique for Editing of Phage Genome
2. CRISPR-Cas System for Editing of Phage Genome
References
- Karam, J.D. Molecular Biology of Bacteriophage T4 Washington; American Society for Microbiology: Washington, DC, USA, 1994.
- Oda, M.; Morita, M.; Unno, H.; Tanji, Y. Rapid detection of Escherichia coli O157:H7 by using green fluorescent protein-labeled PP01 bacteriophage. Appl. Environ. Microbiol. 2004, 70, 527–534.
- Namura, M.; Hijikata, T.; Miyanaga, K.; Tanji, Y. Detection of Escherichia coli with fluorescent labeled phages that have a broad host range to E. coli in sewage water. Biotechnol. Prog. 2008, 24, 481–486.
- Sarkis, G.J.; Jacobs, W.R., Jr.; Hatfull, G.F. L5 luciferase reporter mycobacteriophages: A sensitive tool for the detection and assay of live mycobacteria. Mol. Microbiol. 1995, 15, 1055–1067.
- Rao, V.B.; Mitchell, M.S. The N-terminal ATPase site in the large terminase protein gp17 is critically required for DNA packaging in bacteriophage T4. J. Mol. Biol. 2001, 314, 401–411.
- Marinelli, L.J.; Piuri, M.; Swigonova, Z.; Balachandran, A.; Oldfield, L.M.; Van Kessel, J.C.; Hatfull, G.F. BRED: A simple and powerful tool for constructing mutant and recombinant bacteriophage genomes. PLoS ONE 2008, 3, e3957.
- Thomason, L.C.; Oppenheim, A.B.; Court, D.L. Modifying bacteriophage lambda with recombineering. Methods Mol. Biol. 2009, 501, 239–251.
- Marinelli, L.J.; Hatfull, G.F.; Piuri, M. Recombineering: A powerful tool for modification of bacteriophage genomes. Bacteriophage 2012, 2, 5–14.
- Murphy, K.C. Phage recombinases and their applications. Adv. Virus Res. 2012, 83, 367–414.
- Nafissi, N.; Slavcev, R. Bacteriophage recombination systems and biotechnical applications. Appl. Microbiol. Biotechnol. 2014, 98, 2841–2851.
- Erickson, S.; Paulson, J.; Brown, M.; Hahn, W.; Gil, J.; Barron-Montenegro, R.; Moreno-Switt, A.I.; Eisenberg, M.; & Nguyen, M.M. Isolation and engineering of a Listeria grayi bacteriophage. Sci. Rep. 2021, 11, 18947.
- Masuda, Y.; Kawabata, S.; Uedoi, T.; Honjoh, K.I.; Miyamoto, T. Construction of Leaderless-Bacteriocin-Producing Bacteriophage Targeting, E. coli and Neighboring Gram-Positive Pathogens. Microbiol. Spectr. 2021, 9, e0014121.
- Haft, D.H.; Selengut, J.; Mongodin, E.F.; Nelson, K.E. A Guild of 45 CRISPR-Associated (Cas) Protein Families and Multiple CRISPR/Cas Subtypes Exist in Prokaryotic Genomes. PLoS Comput. Biol. 2005, 1, 60.
- Godde, J.S.; Bickerton, A. The Repetitive DNA Elements Called CRISPRs and Their Associated Genes: Evidence of Horizontal Transfer among Prokaryotes. J. Mol. Evol. 2006, 62, 718–729.
- Barrangou, R.; Fremaux, C.; Deveau, H.; Richards, M.; Boyaval, P.; Moineau, S.; Romero, D.A.; Horvath, P. CRISPR Provides Acquired Resistance against Viruses in Prokaryotes. Science 2007, 315, 1709–1712.
- Hatoum-Aslan, A. Phage Genetic Engineering Using CRISPR⁻Cas Systems. Viruse 2018, 10, 335.
- Koonin, E.V.; Makarova, K.S.; Zhang, F. Diversity, classification and evolution of CRISPR-Cas systems. Curr. Opin. Microbiol. 2017, 37, 67–78.
- Kiro, R.; Shitrit, D.; Qimron, U. Efficient engineering of a bacteriophage genome using the type I-E CRISPR-Cas system. RNA Biol. 2014, 11, 42–44.
- Martel, B.; Moineau, S. CRISPR-Cas: An efficient tool for genome engineering of virulent bacteriophages. Nucleic Acids Res. 2014, 42, 9504–9513.
- Box, A.M.; McGuffie, M.J.; O’Hara, B.J.; Seed, K.D. Functional analysis of bacteriophage immunity through a Type I-E CRISPR-Cas system in Vibrio cholerae and its application in bacteriophage genome engineering. J. Bacteriol. 2015, 198, 578–590.
- Garneau, J.E.; Dupuis, M.E.; Villion, M.; Romero, D.A.; Barrangou, R.; Boyaval, P.; Fremaux, C.; Horvath, P.; Magadan, A.H.; Moineau, S. The CRISPR/Cas bacterial immune system cleaves bacteriophage and plasmid DNA. Nature 2010, 468, 67–71.
- Deltcheva, E.; Chylinski, K.; Sharma, C.M.; Gonzales, K.; Chao, Y.; Pirzada, Z.A.; Eckert, M.R.; Vogel, J.; Charpentier, E. CRISPR RNA maturation by trans-encoded small RNA and host factor RNase III. Nature 2011, 471, 602–607.
- Shah, S.A.; Erdmann, S.; Mojica, F.J.M.; Garrett, R.A. Protospacer recognition motifs: Mixed identities and functional diversity. RNA Biol. 2013, 10, 891–899.
- Hsu, P.D.; Scott, D.A.; Weinstein, J.A.; Ran, F.A.; Konermann, S.; Agarwala, V.; Li, Y.; Fine, E.J.; Wu, X.; Shalem, O.; et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat. Biotechnol. 2013, 31, 827–832.
- Jiang, W.; Bikard, D.; Cox, D.; Zhang, F.; Marraffini, L.A. RNA-guided editing of bacterial genomes using CRISPR-Cas systems. Nat. Biotechnol. 2013, 31, 233–239.
- Mohanraju, P.; Makarova, K.S.; Zetsche, B.; Zhang, F.; Koonin, E.V.; van der Oost, J. Diverse evolutionary roots and mechanistic variations of the CRISPR-Cas systems. Science 2016, 353, 5147.
- Lemay, M.L.; Tremblay, D.M.; Moineau, S. Genome Engineering of Virulent Lactococcal Phages Using CRISPR-Cas9. ACS Synth. Biol. 2017, 21, 1351–1358.
- Tao, P.; Wu, X.; Tang, W.C.; Zhu, J.; Rao, V. Engineering of Bacteriophage T4 Genome Using CRISPR-Cas9. ACS Synth. Biol. 2017, 6, 1952–1961.
- Lee, C.M.; Davis, T.H.; Bao, G. Examination of CRISPR/Cas9 design tools and the effect of target site accessibility on Cas9 activity. Exp. Physiol. 2017, 103, 456–460.
- Mohr, S.E.; Hu, Y.; Ewen-Campen, B.; Housden, B.E.; Viswanatha, R.; Perrimon, N. CRISPR guide RNA design for research applications. FEBS J. 2016, 283, 3232–3238.
- Dang, Y.; Jia, G.; Choi, J.; Ma, H.; Anaya, E.; Ye, C.; Shankar, P.; Wu, H. Optimizing sgRNA structure to improve CRISPR-Cas9 knockout efficiency. Genome Biol. 2015, 16, 280.
- Sharan, S.K.; Thomason, L.C.; Kuznetsov, S.G.; Court, D.L. Recombineering: A homologous recombination-based method of genetic engineering. Nat. Protoc. 2009, 4, 206–223.
- Kosuri, S.; Church, G.M. Large-scale de novo DNA synthesis: Technologies and applications. Nat. Methods 2014, 11, 499–507.
- Hupfeld, M.; Trasanidou, D.; Ramazzini, L.; Klumpp, J.; Loessner, M.J.; Kilcher, S. A functional type II-A CRISPR-Cas system from Listeria enables efficient genome editing of large non-integrating bacteriophage. Nucleic Acids Res. 2018, 46, 6920–6933.
- Bari, S.M.N.; Walker, F.C.; Cater, K.; Aslan, B.; Hatoum-Aslan, A. Strategies for Editing Virulent Staphylococcal Phages Using CRISPR-Cas10. ACS Synth. Biol. 2017, 6, 2316–2325.