The Aβ peptide, consisting of 39–43 amino acids, is derived from the abnormal processing of the amyloid precursor protein (APP), and the accumulation of Aβ peptide has been considered as a hallmark of AD pathogenetic development
[11][32]. The enzymes α-secretase, β-secretase (also called BACE), and γ-secretase take active roles in the processing of APP
[12][33]. Tau proteins within the brain cells of AD brains are misfolded and abnormally shaped, deposits of which form tangles within the neural cells
[13][34]. In AD, it is common to find tau hyperphosphorylation and aggregation, thus losing its ability to maintain the microtubule tracks; as a result, tau dysfunction could lead to the retraction of neuronal processes and thus cell death
[13][34].
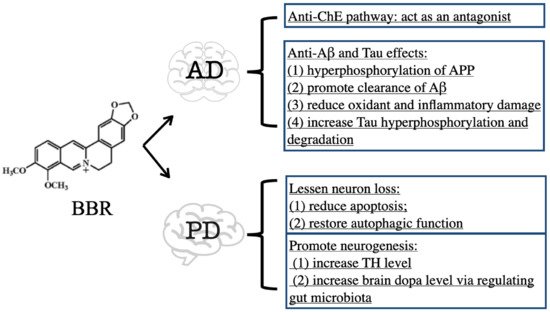
The oral administration of BBR significantly ameliorated learning deficits and spatial memory retention in transgenic mouse models of AD (TgCRND8 mice, APP/PS1 mice, and 3×Tg AD mice)
[14][15][16][17][35,36,37,38]. A mechanistic study showed that BBR significantly decreased the levels of C-terminal fragments of APP and the hyperphosphorylation of APP via the protein kinase B/glycogen synthase kinase 3 (AKT/GSK3) signaling pathway
[14][35]. BBR also inhibited the activity of β/γ-secretases or suppressed PRKR-like endoplasmic reticulum kinase/eukaryotic translation initiation factor-2 α (PERK/EIF2α) signaling-mediated BACE1 translation to downregulate the Aβ level in the AD mouse hippocampus
[15][16][18][36,37,39]. In addition, promoting the clearance of Aβ is another mechanistic aspect of BBR. To promote Aβ clearance, BBR activated the autophagic process through initiating the phosphoinositide 3-kinase (PI3K)/Beclin-1 pathway
[17][38] or by inhibiting the mammalian target of rapamycin/P70 S6 kinase (mTOR/p70S6K) signaling
[19][40]. Additionally, Aβ is toxic to neural cells, as it can cause pore formation resulting in ion leakage, disturb cellular calcium balance, and destroy membrane potential, thus leading to apoptosis, synaptic loss, and cytoskeleton disruption
[20][41]. BBR is effective in preventing Aβ-induced damage to neural cells. Bilaterally injecting rats with Aβ induced learning and memory impairments, while BBR administration ameliorated Aβ-induced toxicity
[21][42]. BBR showed this beneficial effect via modulating the Ca
2+-activated K
+ channel to maintain the optimal level of Ca
2+ entry
[21][42]. Moreover, BBR reduced Aβ-related oxidative and inflammatory damage. The antioxidant effect of BBR was exerted via downregulating reactive oxygen species (ROS) level, promoting the activity of glutathione (GSH), and inhibiting lipid peroxidation
[22][43]. BBR also normalized the production of cytokines such as tumor necrosis factor α (TNFα), interleukin 12 (IL-12), IL-6, and IL-1β to retard inflammation
[9][30]. In addition, exposure to Aβ could potentially lead to microglial activation, thereby triggering a detrimental neural response
[23][44]. No in vivo function of BBR regarding microglial activation has been revealed; only an in vitro study indicated that BBR could inhibit Aβ-induced microglial activation via a silencing of cytokine signaling factor 1 (SOCS1)-dependent modulation of the microglial M1/M2 activated state
[24][45].
Targeting Tau, BBR can reduce its hyperphosphorylation and increase its degradation. In 3×Tg AD mice, BBR improved the spatial learning capacity, memory retention, and the mechanism involved in reducing tau hyperphosphorylation via modulation of the AKT/glycogen synthase kinase 3β (GSK3β) pathway, enhancing autophagic flux, and increasing tau clearance through the PI3K/Beclin-1/B-cell lymphoma 2 (Bcl-2) pathway
[25][46]. In APP/PS1 mice, BBR was found to suppress nuclear factor kappa-light-chain enhancer of the activated B cells (NF-κB) signaling pathway to limit tau hyperphosphorylation
[22][43].
In conclusion, BBR exhibits therapeutic efficacy on PD mainly through the inhibition of ChE activity and suppression of Aβ- and Tau-induced toxicity. The downregulation of ChE activity by BBR contributes to increased ACh availability in the brain
[8][9][29,30]. Both Aβ and Tau are toxic to neural cells via triggering oxidant, inflammatory, and even death signals, while BBR can degrade Aβ and Tau to ameliorate their toxicity
[15][16][17][18][19][21][22][25][36,37,38,39,40,42,43,46] (
Figure 12).