Amyotrophic lateral sclerosis (ALS) is a fatal disease characterized by the degeneration of cortical and spinal motor neurons. With no effective treatment available to date, patients face progressive paralysis and eventually succumb to the disease due to respiratory failure within only a few years. Intriguingly, a key feature present in both ALS patients and rodent models of the disease is cortical hyperexcitability and hyperconnectivity, the mechanisms of which still remain incompletely understood. We here recapitulate current findings arguing for cell autonomous and non-cell autonomous mechanisms causing cortical excitation and inhibition imbalance, which is involved in the degeneration of motor neurons in ALS. Moreover, we will highlight recent evidence that strongly indicate a cardinal role for motor cortex as a main driver and source of the disease, thus arguing for a corticofugal trajectory of the pathology.
- amyotrophic lateral sclerosis
- excitability
- upper motor neurons
- neurodegeneration
- motor cortex
- microcircuit
- interneurons
- astrocytes
1. Introduction
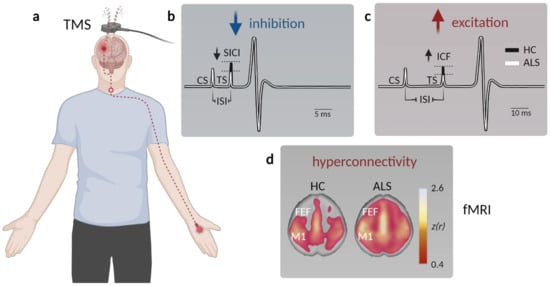
2. Clinical Evidence for Cortical Hyperexcitability in ALS
3. Circuit Mechanisms Involved in Cortical Hyperexcitability
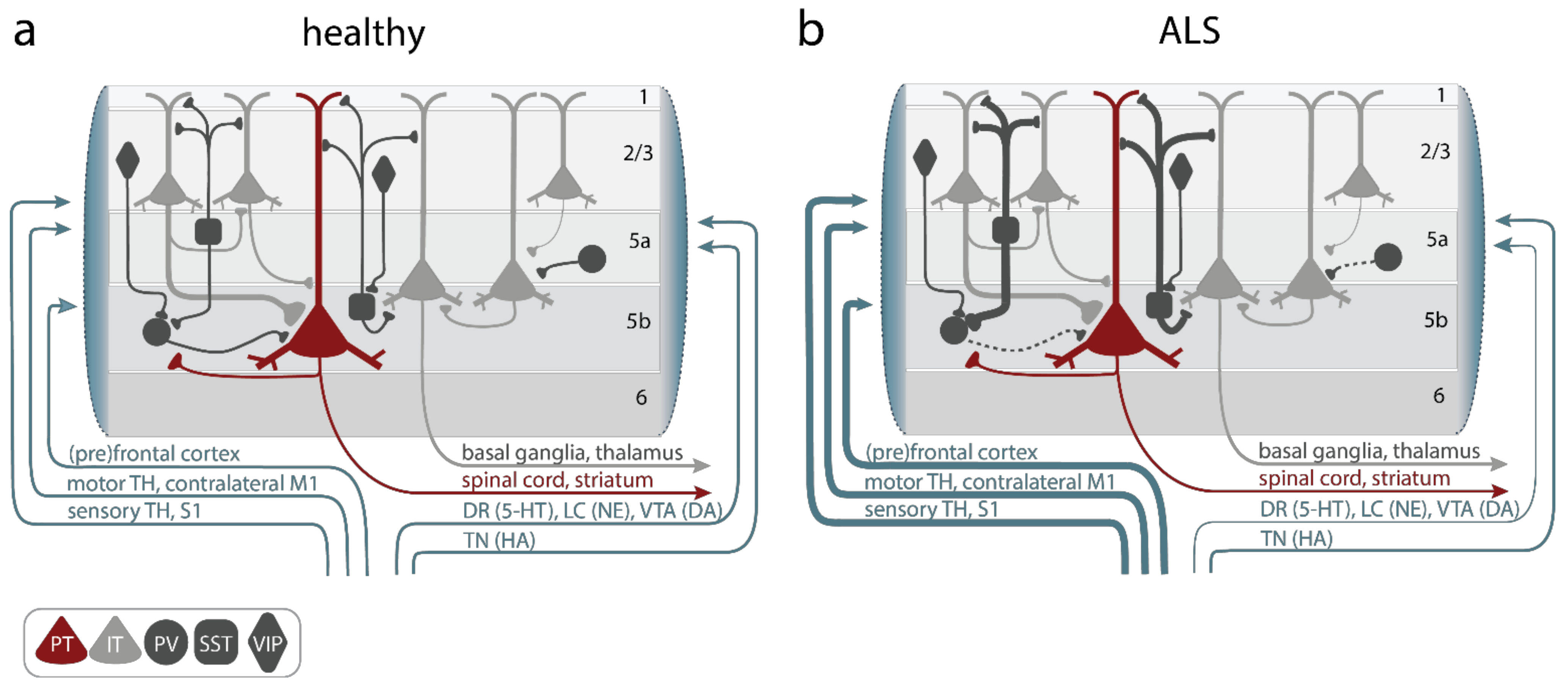
3.1. Alterations of Upper Motor Neurons
3.2. Increased Excitatory Inputs to UMNs
3.3. Reduced Cortical Inhibition
3.4. Altered Neuromodulation
References
- Kiernan, M.C.; Vucic, S.; Cheah, B.C.; Turner, M.R.; Eisen, A.; Hardiman, O.; Burrell, J.R.; Zoing, M.C. Amyotrophic lateral sclerosis. Lancet 2011, 377, 942–955.
- Zhang, W.; Zhang, L.; Liang, B.; Schroeder, D.; Zhang, Z.-W.; Cox, G.A.; Li, Y.; Lin, D.-T. Hyperactive somatostatin interneurons contribute to excitotoxicity in neurodegenerative disorders. Nat. Neurosci. 2016, 19, 557–559.
- Hardiman, O.; Al-Chalabi, A.; Chio, A.; Corr, E.M.; Logroscino, G.; Robberecht, W.; Shaw, P.J.; Simmons, Z.; van den Berg, L.H. Amyotrophic lateral sclerosis. Nat. Rev. Dis. Primers 2017, 3, 17071.
- Chia, R.; Chiò, A.; Traynor, B.J. Novel genes associated with amyotrophic lateral sclerosis: Diagnostic and clinical implications. Lancet Neurol. 2018, 17, 94–102.
- Brown, R.H.; Al-Chalabi, A. Amyotrophic Lateral Sclerosis. N. Engl. J. Med. 2017, 377, 162–172.
- Chen, S.; Sayana, P.; Zhang, X.; Le, W. Genetics of amyotrophic lateral sclerosis: An update. Mol. Neurodegener. 2013, 8, 28.
- Mejzini, R.; Flynn, L.L.; Pitout, I.L.; Fletcher, S.; Wilton, S.D.; Akkari, P.A. ALS Genetics, Mechanisms, and Therapeutics: Where Are We Now? Front. Neurosci. 2019, 13, 1310.
- Taylor, J.P.; Brown, R.H., Jr.; Cleveland, D.W. Decoding ALS: From genes to mechanism. Nature 2016, 539, 197–206.
- Mehta, P.R.; Jones, A.R.; Opie-Martin, S.; Shatunov, A.; Iacoangeli, A.; Al Khleifat, A.; Smith, B.N.; Topp, S.; Morrison, K.E.; Shaw, P.J.; et al. Younger age of onset in familial amyotrophic lateral sclerosis is a result of pathogenic gene variants, rather than ascertainment bias. J. Neurol. Neurosurg. Psychiatry 2018, 90, 268–271.
- Renton, A.E.; Chio, A.; Traynor, B.J. State of play in amyotrophic lateral sclerosis genetics. Nat. Neurosci. 2013, 17, 17–23.
- Ling, S.C.; Polymenidou, M.; Cleveland, D.W. Converging mechanisms in ALS and FTD: Disrupted RNA and protein homeostasis. Neuron 2013, 79, 416–438.
- Neumann, M.; Sampathu Deepak, M.; Kwong Linda, K.; Truax Adam, C.; Micsenyi Matthew, C.; Chou Thomas, T.; Bruce, J.; Schuck, T.; Grossman, M.; Clark Christopher, M.; et al. Ubiquitinated TDP-43 in Frontotemporal Lobar Degeneration and Amyotrophic Lateral Sclerosis. Science 2006, 314, 130–133.
- Mori, K.; Weng, S.-M.; Arzberger, T.; May, S.; Rentzsch, K.; Kremmer, E.; Schmid, B.; Kretzschmar, H.A.; Cruts, M.; Van Broeckhoven, C.; et al. The C9orf72 GGGGCC Repeat Is Translated into Aggregating Dipeptide-Repeat Proteins in FTLD/ALS. Science 2013, 339, 1335–1338.
- Deng, H.-X.; Zhai, H.; Bigio, E.H.; Yan, J.; Fecto, F.; Ajroud, K.; Mishra, M.; Ajroud-Driss, S.; Heller, S.; Sufit, R.; et al. FUS-immunoreactive inclusions are a common feature in sporadic and non-SOD1 familial amyotrophic lateral sclerosis. Ann. Neurol. 2010, 67, 739–748.
- Ikenaka, K.; Ishigaki, S.; Iguchi, Y.; Kawai, K.; Fujioka, Y.; Yokoi, S.; Abdelhamid, R.F.; Nagano, S.; Mochizuki, H.; Katsuno, M.; et al. Characteristic Features of FUS Inclusions in Spinal Motor Neurons of Sporadic Amyotrophic Lateral Sclerosis. J. Neuropathol. Exp. Neurol. 2020, 79, 370–377.
- De Boer, E.M.J.; Orie, V.K.; Williams, T.; Baker, M.R.; De Oliveira, H.M.; Polvikoski, T.; Silsby, M.; Menon, P.; van den Bos, M.; Halliday, G.M.; et al. TDP-43 proteinopathies: A new wave of neurodegenerative diseases. J. Neurol. Neurosurg. Psychiatry 2020, 92, 86–95.
- Benson, B.C.; Shaw, P.J.; Azzouz, M.; Highley, J.R.; Hautbergue, G.M. Proteinopathies as Hallmarks of Impaired Gene Expression, Proteostasis and Mitochondrial Function in Amyotrophic Lateral Sclerosis. Front. Neurosci. 2021, 15, 783624.
- Polymenidou, M.; Lagier-Tourenne, C.; Hutt, K.R.; Bennett, C.F.; Cleveland, D.W.; Yeo, G.W. Misregulated RNA processing in amyotrophic lateral sclerosis. Brain Res. 2012, 1462, 3–15.
- Kawamata, H.; Manfredi, G. Mitochondrial dysfunction and intracellular calcium dysregulation in ALS. Mech. Ageing Dev. 2010, 131, 517–526.
- Barber, S.C.; Shaw, P.J. Oxidative stress in ALS: Key role in motor neuron injury and therapeutic target. Free Radic. Biol. Med. 2010, 48, 629–641.
- Xiao, S.; McLean, J.; Robertson, J. Neuronal intermediate filaments and ALS: A new look at an old question. Biochim. Biophys. Acta BBA—Mol. Basis Dis. 2006, 1762, 1001–1012.
- Marinković, P.; Reuter, M.S.; Brill, M.S.; Godinho, L.; Kerschensteiner, M.; Misgeld, T. Axonal transport deficits and degeneration can evolve independently in mouse models of amyotrophic lateral sclerosis. Proc. Natl. Acad. Sci. USA 2012, 109, 4296–4301.
- Leal, S.S.; Cardoso, I.; Valentine, J.S.; Gomes, C.M. Calcium Ions Promote Superoxide Dismutase 1 (SOD1) Aggregation into Non-fibrillar Amyloid: A Link to Toxic Effects of Calcium Overload in Amyotrophic Lateral Sclerosis (ALS)? J. Biol. Chem. 2013, 288, 25219–25228.
- Lautenschlaeger, J.; Prell, T.; Grosskreutz, J. Endoplasmic reticulum stress and the ER mitochondria calcium cycle in amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. 2012, 13, 166–177.
- Grosskreutz, J.; Bosch, L.V.D.; Keller, B.U. Calcium dysregulation in amyotrophic lateral sclerosis. Cell Calcium 2010, 47, 165–174.
- Dupuis, L.; Pradat, P.-F.; Ludolph, A.C.; Loeffler, J.-P. Energy metabolism in amyotrophic lateral sclerosis. Lancet Neurol. 2011, 10, 75–82.
- Schulthess, I.; Gorges, M.; Müller, H.-P.; Lulé, D.; Del Tredici, K.; Ludolph, A.C.; Kassubek, J. Functional connectivity changes resemble patterns of pTDP-43 pathology in amyotrophic lateral sclerosis. Sci. Rep. 2016, 6, 38391.
- Gunes, Z.I.; Kan, V.W.Y.; Ye, X.; Liebscher, S. Exciting Complexity: The Role of Motor Circuit Elements in ALS Pathophysiology. Front. Neurosci. 2020, 14, 573.
- Gunes, T.; Sirin, N.G.; Sahin, S.; Kose, E.; Isak, B. Use of CMAP, MScan fit-MUNE, and MUNIX in understanding neurodegeneration pattern of ALS and detection of early motor neuron loss in daily practice. Neurosci. Lett. 2020, 741, 135488.
- Dadon-Nachum, M.; Melamed, E.; Offen, D. The “Dying-Back” Phenomenon of Motor Neurons in ALS. J. Mol. Neurosci. 2011, 43, 470–477.
- Eisen, A.; Weber, M. The motor cortex and amyotrophic lateral sclerosis. Muscle Nerve 2001, 24, 564–573.
- Braak, H.; Brettschneider, J.; Ludolph, A.C.; Lee, V.M.; Trojanowski, J.Q.; Del Tredici, K. Amyotrophic lateral sclerosis—A model of corticofugal axonal spread. Nat. Rev. Neurol. 2013, 9, 708–714.
- Baker, M.R. ALS—dying forward, backward or outward? Nat. Rev. Neurol. 2014, 10, 660.
- Grieve, S.M.; Menon, P.; Korgaonkar, M.S.; Gomes, L.; Foster, S.; Kiernan, M.C.; Vucic, S. Potential structural and functional biomarkers of upper motor neuron dysfunction in ALS. Amyotroph. Lateral Scler. Front. Degener. 2015, 17, 85–92.
- Geevasinga, N.; Van den Bos, M.; Menon, P.; Vucic, S. Utility of Transcranial Magnetic Simulation in Studying Upper Motor Neuron Dysfunction in Amyotrophic Lateral Sclerosis. Brain Sci. 2021, 11, 906.
- Vucic, S.; Nicholson, G.A.; Kiernan, M.C. Cortical hyperexcitability may precede the onset of familial amyotrophic lateral sclerosis. Brain 2008, 131, 1540–1550.
- Kujirai, T.; Caramia, M.D.; Rothwell, J.C.; Day, B.L.; Thompson, P.D.; Ferbert, A.; Wroe, S.; Asselman, P.; Marsden, C.D. Corticocortical inhibition in human motor cortex. J. Physiol. 1993, 471, 501–519.
- Vucic, S.; Kiernan, M.C. Axonal excitability properties in amyotrophic lateral sclerosis. Clin. Neurophysiol. 2006, 117, 1458–1466.
- Menke, R.A.L.; Agosta, F.; Grosskreutz, J.; Filippi, M.; Turner, M.R. Neuroimaging Endpoints in Amyotrophic Lateral Sclerosis. Neurotherapeutics 2016, 14, 11–23.
- Vucic, S.; Pavey, N.; Haidar, M.; Turner, B.J.; Kiernan, M.C. Cortical hyperexcitability: Diagnostic and pathogenic biomarker of ALS. Neurosci. Lett. 2021, 759, 136039.
- Vucic, S.; van den Bos, M.; Menon, P.; Howells, J.; Dharmadasa, T.; Kiernan, M.C. Utility of threshold tracking transcranial magnetic stimulation in ALS. Clin. Neurophysiol. Pract. 2018, 3, 164–172.
- McMackin, R.; Dukic, S.; Costello, E.; Pinto-Grau, M.; McManus, L.; Broderick, M.; Chipika, R.; Iyer, P.M.; Heverin, M.; Bede, P.; et al. Cognitive network hyperactivation and motor cortex decline correlate with ALS prognosis. Neurobiol. Aging 2021, 104, 57–70.
- Hallett, M. Transcranial Magnetic Stimulation: A Primer. Neuron 2007, 55, 187–199.
- Triggs, W.J.; Menkes, D.; Onorato, J.; Yan, R.S.H.; Young, M.S.; Newell, K.; Sander, H.W.; Soto, O.; Chiappa, K.H.; Cros, D. Transcranial magnetic stimulation identifies upper motor neuron involvement in motor neuron disease. Neurology 1999, 53, 605.
- Agarwal, S.; Highton-Williamson, E.; Caga, J.; Howells, J.; Dharmadasa, T.; Matamala, J.M.; Ma, Y.; Shibuya, K.; Hodges, J.R.; Ahmed, R.M.; et al. Motor cortical excitability predicts cognitive phenotypes in amyotrophic lateral sclerosis. Sci. Rep. 2021, 11, 2172.
- Zanette, G.; Tamburin, S.; Manganotti, P.; Refatti, N.; Forgione, A.; Rizzuto, N. Changes in motor cortex inhibition over time in patients with amyotrophic lateral sclerosis. J. Neurol. 2002, 249, 1723–1728.
- Mills, K.R. Motor neuron disease. Brain 1995, 118, 971–982.
- Rösler, K.; Truffert, A.; Hess, C.; Magistris, M. Quantification of upper motor neuron loss in amyotrophic lateral sclerosis. Clin. Neurophysiol. 2000, 111, 2208–2218.
- Prout, A.J.; Eisen, A.A. The cortical silent period and amyotrophic lateral sclerosis. Muscle Nerve 1994, 17, 217–223.
- Yokota, T.; Yoshino, A.; Inaba, A.; Saito, Y. Double cortical stimulation in amyotrophic lateral sclerosis. J. Neurol. Neurosurg. Psychiatry 1996, 61, 596–600.
- Ziemann, U.; Winter, M.; Reimers, C.D.; Reimers, K.; Tergau, F.; Paulus, W. Impaired motor cortex inhibition in patients with amyotrophic lateral sclerosis: Evidence from paired transcranial magnetic stimulation. Neurology 1997, 49, 1292–1298.
- Eisen, A.; Weber, M. Neurophysiological evaluation of cortical function in the early diagnosis of ALS. Amyotroph. Lateral Scler. 2000, 1, S47–S51.
- Turner, M.R.; Osei-Lah, A.D.; Hammers, A.; Al-Chalabi, A.; Shaw, C.; Andersen, P.M.; Brooks, D.; Leigh, P.N.; Mills, K.R. Abnormal cortical excitability in sporadic but not homozygous D90A SOD1 ALS. J. Neurol. Neurosurg. Psychiatry 2005, 76, 1279–1285.
- Vucic, S.; Cheah, B.C.; Yiannikas, C.; Kiernan, M.C. Cortical excitability distinguishes ALS from mimic disorders. Clin. Neurophysiol. 2011, 122, 1860–1866.
- Menon, P.; Kiernan, M.C.; Vucic, S. Cortical hyperexcitability precedes lower motor neuron dysfunction in ALS. Clin. Neurophysiol. 2015, 126, 803–809.
- Tankisi, H.; Nielsen, C.S.-Z.; Howells, J.; Cengiz, B.; Samusyte, G.; Koltzenburg, M.; Blicher, J.U.; Møller, A.T.; Pugdahl, K.; Fuglsang-Frederiksen, A.; et al. Early diagnosis of amyotrophic lateral sclerosis by threshold tracking and conventional transcranial magnetic stimulation. Eur. J. Neurol. 2021, 28, 3030–3039.
- Werhahn, K.J.; Kunesch, E.; Noachtar, S.; Benecke, R.; Classen, J. Differential effects on motorcortical inhibition induced by blockade of GABA uptake in humans. J. Physiol. 1999, 517, 591–597.
- Ziemann, U.; Tergau, F.; Wassermann, E.M.; Wischer, S.; Hildebrandt, J.; Paulus, W. Demonstration of facilitatory I wave interaction in the human motor cortex by paired transcranial magnetic stimulation. J. Physiol. 1998, 511, 181–190.
- Ziemann, U.; Reis, J.; Schwenkreis, P.; Rosanova, M.; Strafella, A.; Badawy, R.; Müller-Dahlhaus, F. TMS and drugs revisited 2014. Clin. Neurophysiol. 2015, 126, 1847–1868.
- Menon, P.; Geevasinga, N.; Bos, M.V.D.; Yiannikas, C.; Kiernan, M.C.; Vucic, S. Cortical hyperexcitability and disease spread in amyotrophic lateral sclerosis. Eur. J. Neurol. 2017, 24, 816–824.
- Van den Bos, M.A.J.; Higashihara, M.; Geevasinga, N.; Menon, P.; Kiernan, M.C.; Vucic, S. Imbalance of cortical facilitatory and inhibitory circuits underlies hyperexcitability in ALS. Neurology 2018, 91, e1669–e1676.
- Pfurtscheller, G.; Lopes da Silva, F.H. Event-related EEG/MEG synchronization and desynchronization: Basic principles. Clin. Neurophysiol. 1999, 110, 1842–1857.
- Kasahara, T.; Terasaki, K.; Ogawa, Y.; Ushiba, J.; Aramaki, H.; Masakado, Y. The correlation between motor impairments and event-related desynchronization during motor imagery in ALS patients. BMC Neurosci. 2012, 13, 66.
- Bizovičar, N.; Dreo, J.; Koritnik, B.; Zidar, J. Decreased movement-related beta desynchronization and impaired post-movement beta rebound in amyotrophic lateral sclerosis. Clin. Neurophysiol. 2014, 125, 1689–1699.
- Riva, N.; Falini, A.; Inuggi, A.; Gonzalez-Rosa, J.; Amadio, S.; Cerri, F.; Fazio, R.; Del Carro, U.; Comola, M.; Comi, G.; et al. Cortical activation to voluntary movement in amyotrophic lateral sclerosis is related to corticospinal damage: Electrophysiological evidence. Clin. Neurophysiol. 2012, 123, 1586–1592.
- Proudfoot, M.; Rohenkohl, G.; Quinn, A.; Colclough, G.L.; Wuu, J.; Talbot, K.; Woolrich, M.W.; Benatar, M.; Nobre, A.C.; Turner, M.R. Altered cortical beta-band oscillations reflect motor system degeneration in amyotrophic lateral sclerosis. Hum. Brain Mapp. 2016, 38, 237–254.
- Logothetis, N.K. What we can do and what we cannot do with fMRI. Nature 2008, 453, 869–878.
- Hermes, D.; Nguyen, M.; Winawer, J. Neuronal synchrony and the relation between the blood-oxygen-level dependent response and the local field potential. PLoS Biol. 2017, 15, e2001461.
- Konrad, C.; Jansen, A.; Henningsen, H.; Sommer, J.; Turski, P.A.; Brooks, B.R.; Knecht, S. Subcortical reorganization in amyotrophic lateral sclerosis. Exp. Brain Res. 2006, 172, 361–369.
- Schoenfeld, M.A.; Tempelmann, C.; Gaul, C.; Kühnel, G.R.; Düzel, E.; Hopf, J.-M.; Feistner, H.; Zierz, S.; Heinze, H.-J.; Vielhaber, S. Functional motor compensation in amyotrophic lateral sclerosis. J. Neurol. 2005, 252, 944–952.
- Douaud, G.; Filippini, N.; Knight, S.; Talbot, K.; Turner, M. Integration of structural and functional magnetic resonance imaging in amyotrophic lateral sclerosis. Brain 2011, 134, 3470–3479.
- Agosta, F.; Spinelli, E.G.; Marjanovic, I.V.; Stevic, Z.; Pagani, E.; Valsasina, P.; Salak-Djokic, B.; Jankovic, M.; Lavrnic, D.; Kostic, V.S.; et al. Unraveling ALS due to SOD1 mutation through the combination of brain and cervical cord MRI. Neurology 2018, 90, e707–e716.
- Li, W.; Zhang, J.; Zhou, C.; Hou, W.; Hu, J.; Feng, H.; Zheng, X. Abnormal Functional Connectivity Density in Amyotrophic Lateral Sclerosis. Front. Aging Neurosci. 2018, 10, 215.
- Commisso, B.; Ding, L.; Varadi, K.; Gorges, M.; Bayer, D.; Boeckers, T.M.; Ludolph, A.C.; Kassubek, J.; Müller, O.J.; Roselli, F. Stage-dependent remodeling of projections to motor cortex in ALS mouse model revealed by a new variant retrograde-AAV9. eLife 2018, 7, e36892.
- Proudfoot, M.; Colclough, G.L.; Quinn, A.; Wuu, J.; Talbot, K.; Benatar, M.; Nobre, A.C.; Woolrich, M.W.; Turner, M.R. Increased cerebral functional connectivity in ALS. Neurology 2018, 90, e1418–e1424.
- Foerster, B.R.; Callaghan, B.C.; Petrou, M.; Den, R.A.E.; Chenevert, T.L.; Feldman, E.L. Decreased motor cortex γ-aminobutyric acid in amyotrophic lateral sclerosis. Neurology 2012, 78, 1596–1600.
- Foerster, B.R.; Pomper, M.G.; Callaghan, B.C.; Petrou, M.; Edden, R.; Mohamed, M.; Welsh, R.C.; Carlos, R.C.; Barker, P.B.; Feldman, E. An Imbalance Between Excitatory and Inhibitory Neurotransmitters in Amyotrophic Lateral Sclerosis Revealed by Use of 3-T Proton Magnetic Resonance Spectroscopy. JAMA Neurol. 2013, 70, 1009–1016.
- Petri, S.; Krampfl, K.; Hashemi, F.; Grothe, C.; Hori, A.; Dengler, R.; Bufler, J. Distribution of GABAAReceptor mRNA in the Motor Cortex of ALS Patients. J. Neuropathol. Exp. Neurol. 2003, 62, 1041–1051.
- Aronica, E.; Baas, F.; Iyer, A.; Asbroek, A.L.T.; Morello, G.; Cavallaro, S. Molecular classification of amyotrophic lateral sclerosis by unsupervised clustering of gene expression in motor cortex. Neurobiol. Dis. 2014, 74, 359–376.
- Shepherd, G.M.G. Corticostriatal connectivity and its role in disease. Nat. Rev. Neurosci. 2013, 14, 278–291.
- Nigro, M.J.; Hashikawa-Yamasaki, Y.; Rudy, B. Diversity and Connectivity of Layer 5 Somatostatin-Expressing Interneurons in the Mouse Barrel Cortex. J. Neurosci. 2018, 38, 1622–1633.
- Swanson, O.K.; Maffei, A. From Hiring to Firing: Activation of Inhibitory Neurons and Their Recruitment in Behavior. Front. Mol. Neurosci. 2019, 12, 168.
- Rudy, B.; Fishell, G.; Lee, S.; Hjerling-Leffler, J. Three groups of interneurons account for nearly 100% of neocortical GABAergic neurons. Dev. Neurobiol. 2011, 71, 45–61.
- Tremblay, R.; Lee, S.; Rudy, B. GABAergic interneurons in the neocortex: From cellular properties to circuits. Neuron 2016, 91, 260–292.
- Wood, K.; Blackwell, J.M.; Geffen, M.N. Cortical inhibitory interneurons control sensory processing. Curr. Opin. Neurobiol. 2017, 46, 200–207.
- Lee, S.; Kruglikov, I.; Huang, Z.J.; Fishell, G.; Rudy, B. A disinhibitory circuit mediates motor integration in the somatosensory cortex. Nat. Neurosci. 2013, 16, 1662–1670.
- Yu, J.; Hu, H.; Agmon, A.; Svoboda, K. Recruitment of GABAergic interneurons in the barrel cortex during active tactile behavior. Neuron 2019, 104, 412–427.e414.
- Gu, Q. Neuromodulatory transmitter systems in the cortex and their role in cortical plasticity. Neuroscience 2002, 111, 815–835.
- Conner, J.M.; Kulczycki, M.; Tuszynski, M.H. Unique Contributions of Distinct Cholinergic Projections to Motor Cortical Plasticity and Learning. Cereb. Cortex 2010, 20, 2739–2748.
- Vitrac, C.; Benoit-Marand, M. Monoaminergic Modulation of Motor Cortex Function. Front. Neural Circuits 2017, 11, 72.
- Fogarty, M.J.; Mu, E.W.H.; Noakes, P.; Lavidis, N.A.; Bellingham, M.C. Marked changes in dendritic structure and spine density precede significant neuronal death in vulnerable cortical pyramidal neuron populations in the SOD1G93A mouse model of amyotrophic lateral sclerosis. Acta Neuropathol. Commun. 2016, 4, 77.
- Fogarty, M.J.; Noakes, P.; Bellingham, M.C. Motor Cortex Layer V Pyramidal Neurons Exhibit Dendritic Regression, Spine Loss, and Increased Synaptic Excitation in the Presymptomatic hSOD1G93A Mouse Model of Amyotrophic Lateral Sclerosis. J. Neurosci. 2015, 35, 643–647.
- Fogarty, M.J.; Klenowski, P.M.; Lee, J.D.; Drieberg-Thompson, J.R.; Bartlett, S.E.; Ngo, S.T.; Hilliard, M.A.; Bellingham, M.C.; Noakes, P.G. Cortical synaptic and dendritic spine abnormalities in a presymptomatic TDP-43 model of amyotrophic lateral sclerosis. Sci. Rep. 2016, 6, 37968.
- Kim, J.; Hughes, E.G.; Shetty, A.S.; Arlotta, P.; Goff, L.; Bergles, D.E.; Brown, S.P. Changes in the Excitability of Neocortical Neurons in a Mouse Model of Amyotrophic Lateral Sclerosis Are Not Specific to Corticospinal Neurons and Are Modulated by Advancing Disease. J. Neurosci. 2017, 37, 9037–9053.
- Buskila, Y.; Kékesi, O.; Bellot-Saez, A.; Seah, W.; Berg, T.; Trpceski, M.; Yerbury, J.; Ooi, L. Dynamic interplay between H-current and M-current controls motoneuron hyperexcitability in amyotrophic lateral sclerosis. Cell Death Dis. 2019, 10, 310.
- Saba, L.; Viscomi, M.T.; Martini, A.; Caioli, S.; Mercuri, N.B.; Guatteo, E.; Zona, C. Modified age-dependent expression of NaV1.6 in an ALS model correlates with motor cortex excitability alterations. Neurobiol. Dis. 2019, 130, 104532.
- Dyer, M.S.; Reale, L.A.; Lewis, K.E.; Walker, A.K.; Dickson, T.C.; Woodhouse, A.; Blizzard, C.A. Mislocalisation of TDP-43 to the cytoplasm causes cortical hyperexcitability and reduced excitatory neurotransmission in the motor cortex. J. Neurochem. 2020, 157, 1300–1315.
- Schmitt-John, T.; Drepper, C.; Mußmann, A.; Hahn, P.; Kuhlmann, M.; Thiel, C.; Hafner, M.; Lengeling, A.; Heimann, P.; Jones, J.M.; et al. Mutation of Vps54 causes motor neuron disease and defective spermiogenesis in the wobbler mouse. Nat. Genet. 2005, 37, 1213–1215.
- Nieto-Gonzalez, J.L.; Moser, J.; Lauritzen, M.; Schmitt-John, T.; Jensen, K. Reduced GABAergic Inhibition Explains Cortical Hyperexcitability in the Wobbler Mouse Model of ALS. Cereb. Cortex 2011, 21, 625–635.
- Scekic-Zahirovic, J.; Sanjuan-Ruiz, I.; Kan, V.; Megat, S.; De Rossi, P.; Dieterlé, S.; Cassel, R.; Jamet, M.; Kessler, P.; Wiesner, D.; et al. Cytoplasmic FUS triggers early behavioral alterations linked to cortical neuronal hyperactivity and inhibitory synaptic defects. Nat. Commun. 2021, 12, 3028.
- Khademullah, C.S.; Aqrabawi, A.J.; Place, K.M.; Dargaei, Z.; Liang, X.; Pressey, J.C.; Bedard, S.; Yang, J.W.; Garand, D.; Keramidis, I.; et al. Cortical interneuron-mediated inhibition delays the onset of amyotrophic lateral sclerosis. Brain 2020, 143, 800–810.
- Özdinler, P.H.; Benn, S.; Yamamoto, T.H.; Güzel, M.; Brown, R.H.; Macklis, J.D. Corticospinal Motor Neurons and Related Subcerebral Projection Neurons Undergo Early and Specific Neurodegeneration in hSOD1G93A Transgenic ALS Mice. J. Neurosci. 2011, 31, 4166–4177.
- Clark, R.M.; Blizzard, C.A.; Young, K.M.; King, A.E.; Dickson, T.C. Calretinin and Neuropeptide Y interneurons are differentially altered in the motor cortex of the SOD1G93A mouse model of ALS. Sci. Rep. 2017, 7, 44461.
- Brunet, A.; Stuart-Lopez, G.; Burg, T.; Scekic-Zahirovic, J.; Rouaux, C. Cortical Circuit Dysfunction as a Potential Driver of Amyotrophic Lateral Sclerosis. Front. Neurosci. 2020, 14, 363.
- Takahashi, H.; Snow, B.J.; Bhatt, M.H.; Peppard, R.; Eisen, A.; Calne, D.B. Evidence for a dopaminergic deficit in sporadic amyotrophic lateral sclerosis on positron emission scanning. Lancet 1993, 342, 1016–1018.
- Borasio, G.D.; Linke, R.; Schwarz, J.; Schlamp, V.; Abel, A.; Mozley, P.D.; Tatsch, K. Dopaminergic deficit in amyotrophic lateral sclerosis assessed with IPT single photon emission computed tomography. J. Neurol. Neurosurg. Psychiatry 1998, 65, 263–265.
- Kostic, V.; Gurney, M.E.; Deng, H.-X.; Siddique, T.; Epstein, C.J.; Przedborski, S. Midbrain dopaminergic neuronal degeneration in a transgenic mouse model of familial amyotrophic lateral sclerosis. Ann. Neurol. 1997, 41, 497–504.
- Dentel, C.; Palamiuc, L.; Henriques, A.; Lannes, B.; Spreux-Varoquaux, O.; Gutknecht, L.; René, F.; Echaniz-Laguna, A.; De Aguilar, J.-L.G.; Lesch, K.P.; et al. Degeneration of serotonergic neurons in amyotrophic lateral sclerosis: A link to spasticity. Brain 2013, 136, 483–493.