Psoriasis is an autoimmune disease mediated by disturbed T cells and other immune cells, and is defined by deep-red, well-demarcated skin lesions. Using biologics to target specific immune components, such as upregulated cytokines secreted by activated immune cells, is the most advanced therapy for psoriasis to date.
- psoriasis
- cytokines
- inhibitors
- biologics
- autoimmunity
- immune cells
1. Pathogenesis of Psoriasis
The sustained inflammation that leads to uncontrolled keratinocyte proliferation and faulty differentiation are the hallmarks of psoriasis. Multiple triggers could be from exogenous sources, for instance, infection, skin trauma, smoking habits, drugs, infections and occupational hazards [5]. A strong familial hereditary association of psoriasis-susceptible loci
The sustained inflammation that leads to uncontrolled keratinocyte proliferation and faulty differentiation are the hallmarks of psoriasis. Multiple triggers could be from exogenous sources, for instance, infection, skin trauma, smoking habits, drugs, infections and occupational hazards [1]. A strong familial hereditary association of psoriasis-susceptible loci
PSORS is also a cause of severe psoriasis development which can be detected at an early age [23,24]. If the disease is considered to be acquired, it might be from certain intrinsic conditions, such as hypertension, diabetes mellitus and predisposing metabolic syndrome [5,25].
is also a cause of severe psoriasis development which can be detected at an early age [2][3]. If the disease is considered to be acquired, it might be from certain intrinsic conditions, such as hypertension, diabetes mellitus and predisposing metabolic syndrome [1][4].
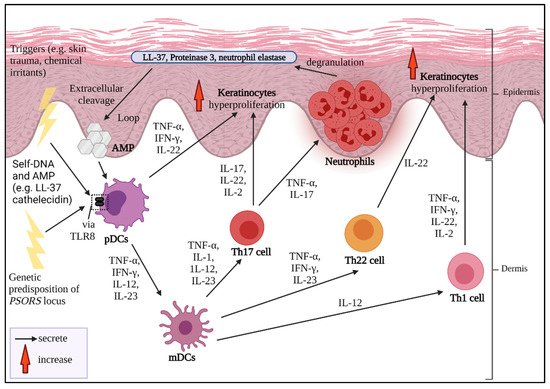
2. Psoriasis and Cytokines as Biologics Target
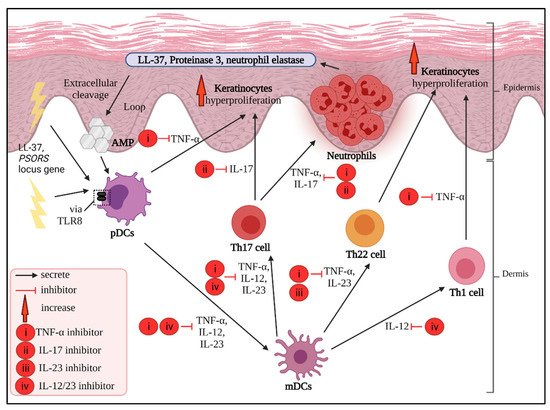
3. Main Potential Cytokine Targets in Psoriasis
3.1. TNF-α Inhibitors
3.2. IL-17 Inhibitors
3.3. IL-23 Inhibitors
3.4. IL-12/23 Inhibitors
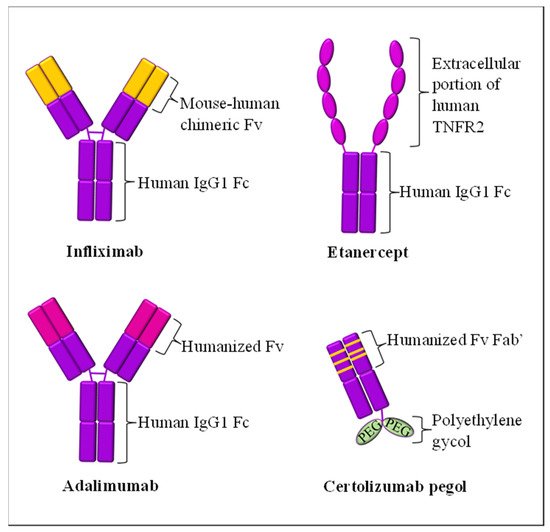
| Cytokine Targets | Biologic Drug Name (Brand) | Year of FDA Approval for Psoriasis Treatment | Molecular Structure | Mode of Action | Possible Side Effects | References |
|---|---|---|---|---|---|---|
| TNF-α | Infliximab (Remicade®) | 2006 | Human-mouse chimeric combination of mAb IgG1 | Inhibit circulating and transmembrane-bound TNF-α | Upper respiratory tract infection, hepatotoxicity, tuberculosis risk, worsening psoriasis | [63,65,66][44][46][47] |
| Etanercept (Enbrel®) | 2004 | Extracellular region of TNFR2 fusion with humanized mAb IgG1 | Inhibit soluble and non-membrane-bound circulatory TNF-α from binding to TNFR2 receptor | Upper and lower respiratory tract infections, rhinitis, pharyngitis, tuberculosis risk | [60,61][41][42] | |
| Adalimumab (Humira®) | 2008 | Humanized mAb IgG1 | Inhibit circulating and transmembrane-bound TNF-α | Upper respiratory tract infection, sinusitis, urinary tract infection | [64,69][45][50] | |
| Golimumab (Simponi®) | Not applicable * | Humanized mAb IgG1κ | Inhibit circulating and transmembrane-bound TNF-α | Recurring psoriasis flare | [72][53] | |
| Certolizumab pegol (Cimzia®) | Not applicable * | Humanized Fab subunit to mAb fusion, with Fc-free PEGylation and no Fc region | Inhibit circulating and transmembrane-bound TNF-α | Urinary tract infections, gastroenteritis, nasopharyngitis, headache, pruritus, tuberculosis risk | [75][56] | |
| IL-17 | Secukinumab (Cosentyx®) | 2015 | Humanized mAb IgG1 | Inhibit IL-17A and IL-17F | Nasopharyngitis, diarrhea, mucocutaneous candidiasis, upper respiratory tract infection, neutropenia | [114,117][96][99] |
| Ixekizumab (Taltz®) | 2016 | Humanized mAb IgG4 | Inhibit IL-17A | Candidiasis, irritable bowel syndrome, neutropenia | [118][100] | |
| Brodalumab (Siliq®) | 2017 | Humanized mAb IgG2 | Block IL-17A and IL-17C receptors | Arthralgia, headaches, fatigue | [122,124,125][104][106][107] | |
| IL-23 | Tildrakizumab (Ilumya®) | 2018 | Humanized mAb IgG1κ | Inhibit IL-23 alpha subunit; p19 subunit | Inflammatory bowel syndrome, acute myocardial infarction | [122,136][104][118] |
| Guselkumab (Tremfya®) | 2017 | Humanized mAb IgG1λ | Inhibit IL-23 alpha subunit; p19 subunit | Upper respiratory tract, nasopharyngitis, headaches, infection | [122,136][104][118] | |
| Risankizumab (Skyrizi®) | 2019 | Humanized mAb IgG1 | Inhibit IL-23A | Nasopharyngitis, upper respiratory tract infection, headache, arthralgia, back pain, diarrhea |
[122,136][104][118] | |
| IL-12/23 | Ustekinumab (Stelara®) | 2009 | Humanized mAb IgG1 | Simultaneously inhibit p40 subunit of IL-12 and IL-23 | Tuberculosis risk | [140,145][122][127] |
References
- Huang, T.-H.; Lin, C.-F.; Alalaiwe, A.; Yang, S.-C.; Fang, J.-Y. Apoptotic or antiproliferative activity of natural products against keratinocytes for the treatment of psoriasis. Int. J. Mol. Sci. 2019, 20, 2558.
- Dhar, S.; Banerjee, R.; Agrawal, N.; Chatterjee, S.; Malakar, R. Psoriasis in children: An insight. Indian J. Dermatol. 2011, 56, 262.
- Rakhshan, A.; Zarrinpour, N.; Moradi, A.; Ahadi, M.; Omrani, M.D.; Ghafouri-Fard, S.; Taheri, M. Genetic variants within ANRIL (antisense non coding RNA in the INK4 locus) are associated with risk of psoriasis. Int. Immunopharmacol. 2020, 78, 106053.
- Kara Polat, A.; Oguz Topal, I.; Karadag, A.S.; Aksoy, H.; Koku Aksu, A.E.; Ozkur, E.; Akbulut, T.O.; Demir, F.T.; Engin, B.; Uzuncakmak, T.K.; et al. The impact of COVID-19 in patients with psoriasis: A multicenter study in Istanbul. Dermatol. Ther. 2021, 34, e14691.
- Campanati, A.; Marani, A.; Martina, E.; Diotallevi, F.; Radi, G.; Offidani, A. Psoriasis as an immune-mediated and inflammatory systemic disease: From pathophysiology to novel therapeutic approaches. Biomedicines 2021, 9, 1511.
- Lew, W.; Bowcock, A.M.; Krueger, J.G. Psoriasis vulgaris: Cutaneous lymphoid tissue supports T-cell activation and ‘Type 1’inflammatory gene expression. Trends Immunol. 2004, 25, 295–305.
- Schon, M.; Behmenburg, C.; Denzer, D.; Schon, M.P. Pathogenic function of IL-1beta in psoriasiform skin lesions of flaky skin (fsn/fsn) mice. Clin. Exp. Immunol. 2001, 123, 505–510.
- Zhu, J.; Paul, W.E. Heterogeneity and plasticity of T helper cells. Cell Res. 2010, 20, 4–12.
- Chiricozzi, A.; Guttman-Yassky, E.; Suárez-Farinas, M.; Nograles, K.E.; Tian, S.; Cardinale, I.; Chimenti, S.; Krueger, J.G. Integrative responses to IL-17 and TNF-α in human keratinocytes account for key inflammatory pathogenic circuits in psoriasis. J. Investig. Dermatol. 2011, 131, 677–687.
- Saeki, H.; Imafuku, S.; Abe, M.; Shintani, Y.; Onozuka, D.; Hagihara, A.; Katoh, N.; Murota, H.; Takeuchi, S.; Sugaya, M.; et al. Poor adherence to medication as assessed by the Morisky Medication Adherence Scale-8 and low satisfaction with treatment in 237 psoriasis patients. J. Dermatol. 2015, 42, 367–372.
- Nast, A.; Jacobs, A.; Rosumeck, S.; Werner, R.N. Efficacy and safety of systemic long-term treatments for moderate-to-severe psoriasis: A systematic review and meta-analysis. J. Investig. Dermatol. 2015, 135, 2641–2648.
- Kaushik, S.B.; Lebwohl, M.G. Review of safety and efficacy of approved systemic psoriasis therapies. Int. J. Dermatol. 2019, 58, 649–658.
- Torres, T.; Filipe, P. Small molecules in the treatment of psoriasis. Drug Dev. Res. 2015, 76, 215–227.
- Gisondi, P.; Del Giglio, M.; Girolomoni, G. Treatment approaches to moderate to severe psoriasis. Int. J. Mol. Sci. 2017, 18, 2427.
- Rønholt, K.; Iversen, L. Old and new biological therapies for psoriasis. Int. J. Mol. Sci. 2017, 18, 2297.
- Ortonne, J.P.; Prinz, J.C. Alefacept: A novel and selective biologic agent for the treatment of chronic plaque psoriasis. Eur. J. Dermatol. 2004, 14, 41–45.
- Liu, C.M.; McKenna, J.K.; Krueger, G.G. Alefacept: A novel biologic in the treatment of psoriasis. Drugs Today 2004, 40, 961–974.
- Langley, R.G.; Cherman, A.M.; Gupta, A.K. Alefacept: An expert review concerning the treatment of psoriasis. Expert Opin. Pharmacother. 2005, 6, 2327–2333.
- Jenneck, C.; Novak, N. The safety and efficacy of alefacept in the treatment of chronic plaque psoriasis. Ther. Clin. Risk Manag. 2007, 3, 411–420.
- Lebwohl, M. Psoriasis. Lancet 2003, 361, 1197–1204.
- Sivamani, R.K.; Correa, G.; Ono, Y.; Bowen, M.P.; Raychaudhuri, S.P.; Maverakis, E. Biological therapy of psoriasis. Indian J. Dermatol. 2010, 55, 161.
- Baliwag, J.; Barnes, D.H.; Johnston, A. Cytokines in psoriasis. Cytokine 2015, 73, 342–350.
- Bak, R.O.; Mikkelsen, J.G. Regulation of cytokines by small RNAs during skin inflammation. J. Biomed. Sci. 2010, 17, 53.
- Wang, Z.; Zheng, H.; Zhou, H.; Huang, N.; Wei, X.; Liu, X.; Teng, X.; Hu, Z.; Zhang, J.; Zhou, X.; et al. Systematic screening and identification of novel psoriasis-specific genes from the transcriptome of psoriasis-like keratinocytes. Mol. Med. Rep. 2019, 19, 1529–1542.
- Gottlieb, A.B.; Chamian, F.; Masud, S.; Cardinale, I.; Abello, M.V.; Lowes, M.A.; Chen, F.; Magliocco, M.; Krueger, J.G. TNF inhibition rapidly down-regulates multiple proinflammatory pathways in psoriasis plaques. J. Immunol. 2005, 175, 2721–2729.
- Grine, L.; Dejager, L.; Libert, C.; Vandenbroucke, R.E. An inflammatory triangle in psoriasis: TNF, type I IFNs and IL-17. Cytokine Growth Factor Rev. 2015, 26, 25–33.
- Mylonas, A.; Conrad, C. Psoriasis: Classical vs. paradoxical. the yin-yang of TNF and Type I interferon. Front. Immunol. 2018, 9, 2746.
- Leon, A.; Liao, W.J.; Gupta, R.; Koo, J.Y.; Wu, J.J. Tumor necrosis factor-α triad: Psoriasis, cardiovascular disease, and depression. Psoriasis Forum 2013, 19, 41–49.
- Calzascia, T.; Pellegrini, M.; Hall, H.; Sabbagh, L.; Ono, N.; Elford, A.R.; Ohashi, P.S. TNF-α is critical for antitumor but not antiviral T cell immunity in mice. J. Clin. Investig. 2007, 117, 3833–3845.
- Chen, X.; Hamano, R.; Subleski, J.J.; Hurwitz, A.A.; Howard, O.Z.; Oppenheim, J.J. Expression of costimulatory TNFR2 induces resistance of CD4+ FoxP3− conventional T cells to suppression by CD4+ FoxP3+ regulatory T cells. J. Immunol. 2010, 185, 174–182.
- Prieto-Perez, R.; Cabaleiro, T.; Dauden, E.; Abad-Santos, F. Gene polymorphisms that can predict response to anti-TNF therapy in patients with psoriasis and related autoimmune diseases. Pharm. J. 2013, 13, 297–305.
- Zhuang, L.; Ma, W.; Cai, D.; Zhong, H.; Sun, Q. Associations between tumor necrosis factor-a polymorphisms and risk of psoriasis: A meta-analysis. PLoS ONE 2013, 8, e68827.
- Murdaca, G.; Gulli, R.; Spano, F.; Lantieri, F.; Burlando, M.; Parodi, A.; Mandich, P.; Puppo, F. TNF-α gene polymorphisms: Association with disease susceptibility and response to anti-TNF-α treatment in psoriatic arthritis. J. Investig. Dermatol. 2014, 134, 2503–2509.
- Mazloom, S.E.; Yan, D.; Hu, J.Z.; Ya, J.; Husni, M.E.; Warren, C.B.; Fernandez, A.P. TNF-α inhibitor–induced psoriasis: A decade of experience at the Cleveland Clinic. JAAD 2020, 83, 1590–1598.
- Ruano, J.; Suárez-Fariñas, M.; Shemer, A.; Oliva, M.; Guttman-Yassky, E.; Krueger, J.G. Molecular and cellular profiling of scalp psoriasis reveals differences and similarities compared to skin psoriasis. PLoS ONE 2016, 11, e0148450.
- Tsoi, L.C.; Spain, S.L.; Ellinghaus, E.; Stuart, P.E.; Capon, F.; Knight, J.; Tejasvi, T.; Kang, H.M.; Allen, M.H.; Lambert, S.; et al. Enhanced meta-analysis and replication studies identify five new psoriasis susceptibility loci. Nat. Commun. 2015, 6, 7001.
- Johnston, A.; Fritz, Y.; Dawes, S.M.; Diaconu, D.; Al-Attar, P.M.; Guzman, A.M.; Chen, C.S.; Fu, W.; Gudjonsson, J.E.; McCormick, T.S.; et al. Keratinocyte overexpression of IL-17C promotes psoriasiform skin inflammation. J. Immunol. 2013, 190, 2252–2262.
- Banno, T.; Gazel, A.; Blumenberg, M. Effects of tumor necrosis factor-α (TNFα) in epidermal keratinocytes revealed using global transcriptional profiling. J. Biol. Chem. 2004, 279, 32633–32642.
- Yost, J.; Gudjonsson, J.E. The role of TNF inhibitors in psoriasis therapy: New implications for associated comorbidities. Med. Rep. 2009, 1, 30.
- Brownstone, N.D.; Hong, J.; Mosca, M.; Hadeler, E.; Liao, W.; Bhutani, T.; Koo, J. Biologic treatments of psoriasis: An update for the clinician. Biol. Targets Ther. 2019, 15, 39.
- Nguyen, T.U.; Koo, J. Etanercept in the treatment of plaque psoriasis. Clinical, cosmetic and investigational dermatology. Clin. Cosmet. Investig. 2009, 19, 77–84.
- Sedger, L.M.; McDermott, M.F. TNF and TNF-receptors: From mediators of cell death and inflammation to therapeutic giants–past, present and future. Cytokine Growth Factor Rev. 2014, 25, 453–472.
- Kivelevitch, D.; Mansouri, B.; Menter, A. Long term efficacy and safety of etanercept in the treatment of psoriasis and psoriatic arthritis. Biol. Targets Ther. 2014, 8, 169.
- Knight, D.M.; Trinh, H.; Le, J.; Siegel, S.; Shealy, D.; McDonough, M.; Ghrayeb, J. Construction and initial characterization of a mouse-human chimeric anti-TNF antibody. Mol. Immunol. 1993, 30, 1443–1453.
- Kaymakcalan, Z.; Sakorafas, P.; Bose, S.; Scesney, S.; Xiong, L.; Hanzatian, D.K.; Salfeld, J.; Sasso, E.H. Comparisons of affinities, avidities, and complement activation of adalimumab, infliximab, and etanercept in binding to soluble and membrane tumor necrosis factor. Clin. Immunol. 2009, 131, 308–316.
- Talbot, C.; Sagar, P.M.; Johnston, M.J.; Finan, P.J.; Burke, D. Infliximab in the surgical management of complex fistulating anal Crohn’s disease. Int. J. Colorectal Dis. 2005, 7, 164–168.
- Tracey, D.; Klareskog, L.; Sasso, E.H.; Salfeld, J.G.; Tak, P.P. Tumor necrosis factor antagonist mechanisms of action: A comprehensive review. Pharmacol. Ther. 2008, 117, 244–279.
- Antoni, C.; Krueger, G.G.; de Vlam, K.; Birbara, C.; Beutler, A.; Guzzo, C.; Zhou, B.; Dooley, L.T.; Kavanaugh, A. Infliximab improves signs and symptoms of psoriatic arthritis: Results of the IMPACT 2 trial. Ann. Rheum. Dis. 2005, 64, 1150–1157.
- Subedi, S.; Gong, Y.; Chen, Y.; Shi, Y. Infliximab and biosimilar infliximab in psoriasis: Efficacy, loss of efficacy, and adverse events. Drug Des. Devel. Ther. 2019, 13, 2491–2502.
- Mazza, J.; Rossi, A.; Weinberg, J.M. Innovative uses of tumor necrosis factor α inhibitors. Dermatol. Clin. 2010, 28, 559–575.
- Chiricozzi, A.; Zangrilli, A.; Bavetta, M.; Bianchi, L.; Chimenti, S.; Saraceno, R. Real-life 9-year experience with adalimumab in psoriasis and psoriatic arthritis: Results of a single-centre, retrospective study. J. Eur. Acad. Dermatol. 2017, 31, 304–311.
- Kamata, M.; Tada, Y. Efficacy and safety of biologics for psoriasis and psoriatic arthritis and their impact on comorbidities: A literature review. Int. J. Mol. Sci. 2020, 21, 1690.
- Xu, Z.; Vu, T.; Lee, H.; Hu, C.; Ling, J.; Yan, H.; Baker, D.; Beutler, A.; Pendley, C.; Wagner, C.; et al. Population pharmacokinetics of golimumab, an anti-tumor necrosis factor-α human monoclonal antibody, in patients with psoriatic arthritis. J. Clin. Pharmacol. 2009, 49, 1056–1070.
- Shealy, D.J.; Cai, A.; Staquet, K.; Baker, A.; Lacy, E.R.; Johns, L.; Vafa, O.; Gunn, G.; Tam, S.; Sague, S.; et al. Characterization of golimumab, a human monoclonal antibody specific for human tumor necrosis factor α. MAbs Taylor Fr. 2010, 2, 428–439.
- Reich, K.; Burden, A.D.; Eaton, J.N.; Hawkins, N.S. Efficacy of biologics in the treatment of moderate to severe psoriasis: A network meta-analysis of randomized controlled trials. Br. J. Dermatol. 2012, 166, 179–188.
- Mariette, X.; Förger, F.; Abraham, B.; Flynn, A.D.; Moltó, A.; Flipo, R.M.; van Tubergen, A.; Shaughnessy, L.; Simpson, J.; Teil, M.; et al. Lack of placental transfer of certolizumab pegol during pregnancy: Results from CRIB, a prospective, postmarketing, pharmacokinetic study. ARD 2018, 77, 228–233.
- Nesbitt, A.; Fossati, G.; Bergin, M.; Stephens, P.; Stephens, S.; Foulkes, R.; Brown, D.; Robinson, M.; Bourne, T. Mechanism of action of certolizumab pegol (CDP870): In vitro comparison with other anti-tumor necrosis factor α agents. Inflamm. Bowel Dis. 2007, 13, 1323–1332.
- Garcia, V.R.; Burls, A.; Cabello, J.B.; Casasempere, P.V.; Bort-Marti, S.; Bernal, J.A. Certolizumab pegol (CDP870) for rheumatoid arthritis in adults. Cochrane Database Syst. Rev. 2017, 9, CD007649.
- Esposito, M.; Carubbi, F.; Giunta, A.; Alunno, A.; Giacomelli, R.; Fargnoli, M.C. Certolizumab pegol for the treatment of psoriatic arthritis and plaque psoriasis. Expert Rev. Clin. Immunol. 2020, 16, 119–128.
- Langley, R.G.; Elewski, B.E.; Lebwohl, M.; Reich, K.; Griffiths, C.E.M.; Papp, K.; Puig, L.; Nakagawa, H.; Spelman, L.; Sigurgeirsson, B.; et al. Secukinumab in plaque psoriasis—Results of two phase 3 trials. NEJM 2014, 371, 326–338.
- Malakouti, M.; Brown, G.E.; Wang, E.; Koo, J.; Levin, E.C. The role of IL-17 in psoriasis. J. Dermatol. Treat. 2015, 26, 41–44.
- Li, H.; Chen, J.; Huang, A.; Stinson, J.; Heldens, S.; Foster, J.; Dowd, P.; Gurney, A.L.; Wood, W.I. Cloning and characterization of IL-17B and IL-17C, two new members of the IL-17 cytokine family. Proc. Natl. Acad. Sci. USA 2000, 97, 773–778.
- Fort, M.M.; Cheung, J.; Yen, D.; Li, J.; Zurawski, S.M.; Lo, S.; Menon, S.; Clifford, T.; Hunte, B.; Lesley, R.; et al. IL-25 induces IL-4, IL-5, and IL-13 and Th2-associated pathologies in vivo. Immunity 2001, 15, 985–995.
- Hymowitz, S.G.; Filvaroff, E.H.; Yin, J.; Lee, J.; Cai, L.; Risser, P.; Maruoka, M.; Mao, W.; Foster, J.; Kelley, R.F.; et al. IL-17s adopt a cystine knot fold: Structure and activity of a novel cytokine, IL-17F, and implications for receptor binding. EMBO J. 2001, 20, 5332–5341.
- Gaffen, S.L. Recent advances in the IL-17 cytokine family. Curr. Opin. 2011, 23, 613–619.
- Frieder, J.; Kivelevitch, D.; Menter, A. Secukinumab: A review of the anti-IL-17A biologic for the treatment of psoriasis. TACD 2018, 9, 5–21.
- Wang, M.; Zhang, S.; Zheng, G.; Huang, J.; Songyang, Z.; Zhao, X.; Lin, X. Gain-of-function mutation of CARD14 leads to spontaneous psoriasis-like skin inflammation through enhanced keratinocyte response to IL-17A. Immunity 2018, 49, 66–79.
- Erbel, C.; Akhavanpoor, M.; Okuyucu, D.; Wangler, S.; Dietz, A.; Zhao, L.; Stellos, K.; Little, K.M.; Lasitschka, F.; Doesch, A.; et al. IL-17A influences essential functions of the monocyte/macrophage lineage and is involved in advanced murine and human atherosclerosis. J. Immunol. 2014, 193, 4344–4355.
- Von Stebut, E.; Boehncke, W.H.; Ghoreschi, K.; Gori, T.; Kaya, Z.; Thaci, D.; Schäffler, A. IL-17A in psoriasis and beyond: Cardiovascular and metabolic implications. Front. Immunol. 2020, 10, 3096.
- Martin, D.A.; Towne, J.E.; Kricorian, G.; Klekotka, P.; Gudjonsson, J.E.; Krueger, J.G.; Russell, C.B. The emerging role of IL-17 in the pathogenesis of psoriasis: Preclinical and clinical findings. J. Investig. Dermatol. 2013, 133, 17–26.
- Gordon, K.B.; Blauvelt, A.; Papp, K.A.; Langley, R.G.; Luger, T.; Ohtsuki, M.; Reich, K.; Amato, D.; Ball, S.G.; Braun, D.K.; et al. Phase 3 trials of ixekizumab in moderate-to-severe plaque psoriasis. NEJM 2016, 375, 345–356.
- Huang, X.D.; Zhang, H.; He, M.X. Comparative and evolutionary analysis of the interleukin 17 gene family in invertebrates. PLoS ONE 2015, 10, e0132802.
- Angkasekwinai, P.; Park, H.; Wang, Y.H.; Wang, Y.H.; Chang, S.H.; Corry, D.B.; Liu, Y.J.; Zhu, Z.; Dong, C. Interleukin 25 promotes the initiation of proallergic type 2 responses. Exp. Med. 2007, 204, 1509–1517.
- Wasilewska, A.; Winiarska, M.; Olszewska, M.; Rudnicka, L. Interleukin-17 inhibitors. A new era in treatment of psoriasis and other skin diseases. Postepy Dermatol Alergol. 2016, 33, 247.
- Krueger, J.G.; Fretzin, S.; Suárez-Fariñas, M.; Haslett, P.A.; Phipps, K.M.; Cameron, G.S.; McColm, J.; Katcherian, A.; Cueto, I.; White, T.; et al. IL-17A is essential for cell activation and inflammatory gene circuits in subjects with psoriasis. J. Allergy Clin. Immunol. 2012, 130, 145–154.
- Kirkham, B.W.; Kavanaugh, A.; Reich, K. Interleukin-17A: A unique pathway in immune-mediated diseases: Psoriasis, psoriatic arthritis and rheumatoid arthritis. Immunology 2014, 141, 133–142.
- Hijnen, D.; Knol, E.F.; Gent, Y.Y.; Giovannone, B.; Beijn, S.J.; Kupper, T.S.; Bruijnzeel-Koomen, C.A.F.M.; Clark, R.A. CD8+ T cells in the lesional skin of atopic dermatitis and psoriasis patients are an important source of IFN-γ, IL-13, IL-17, and IL-22. J. Investig. Dermatol. 2013, 133, 973–979.
- Slominski, A.T.; Hardeland, R.; Zmijewski, M.A.; Slominski, R.M.; Reiter, R.J.; Paus, R. Melatonin: A cutaneous perspective on its production, metabolism, and functions. J. Investig. Dermatol. 2018, 138, 490–499.
- Starnes, T.; Broxmeyer, H.E.; Robertson, M.J.; Hromas, R. Cutting edge: IL-17D, a novel member of the IL-17 family, stimulates cytokine production and inhibits hemopoiesis. J. Immunol. 2002, 2169, 642–646.
- Miossec, P.; Kolls, J.K. Targeting IL-17 and Th 17 cells in chronic inflammation. Nat. Rev. Drug Discov. 2012, 11, 763–776.
- Ruddy, M.J.; Wong, G.C.; Liu, X.K.; Yamamoto, H.; Kasayama, S.; Kirkwood, K.L.; Gaffen, S.L. Functional cooperation between interleukin-17 and tumor necrosis factor-α is mediated by CCAAT/enhancer-binding protein family members. J. Biol. Chem. 2004, 279, 2559–2567.
- Laan, M.; Cui, Z.H.; Hoshino, H.; Lötvall, J.; Sjöstrand, M.; Gruenert, D.C.; Skoogh, B.E.; Lindén, A. Neutrophil recruitment by human IL-17 via CXC chemokine release in the airways. J. Immunol. 1999, 162, 2347–2352.
- Moseley, T.A.; Haudenschild, D.R.; Rose, L.; Reddi, A.H. Interleukin-17 family and IL-17 receptors. Cytokine Growth Factor Rev. 2003, 14, 155–174.
- Harper, E.G.; Guo, C.; Rizzo, H.; Lillis, J.V.; Kurtz, S.E.; Skorcheva, I.; Purdy, D.; Fitch, E.; Iordanov, M.; Blauvelt, A. Th17 cytokines stimulate CCL20 expression in keratinocytes in vitro and in vivo: Implications for psoriasis pathogenesis. J. Investig. Dermatol. 2009, 129, 2175–2183.
- Johansen, C.; Usher, P.A.; Kjellerup, R.B.; Lundsgaard, D.; Iversen, L.; Kragballe, K. Characterization of the interleukin-17 isoforms and receptors in lesional psoriatic skin. Br. J. Dermatol. Suppl. 2009, 160, 319–324.
- Yilmaz, S.B.; Cicek, N.; Coskun, M.; Yegin, O.; Alpsoy, E. Serum and tissue levels of IL-17 in different clinical subtypes of psoriasis. Arch. Dermatol. 2012, 304, 465–469.
- Gaffen, S.L. Structure and signalling in the IL-17 receptor family. Nat. Rev. Immunol. 2009, 9, 556–567.
- Fujishima, S.; Watanabe, H.; Kawaguchi, M.; Suzuki, T.; Matsukura, S.; Homma, T.; Howell, B.G.; Hizawa, N.; Mitsuya, T.; Huang, S.K.; et al. Involvement of IL-17F via the induction of IL-6 in psoriasis. Arch. Dermatol. 2010, 302, 499–505.
- Oliveira, M.D.F.S.P.D.; Rocha, B.D.O.; Duarte, G.V. Psoriasis: Classical and emerging comorbidities. An. Bras. Dermatol. 2015, 90, 9–20.
- Pantelyushin, S.; Haak, S.; Ingold, B.; Kulig, P.; Heppner, F.L.; Navarini, A.A.; Becher, B. Rorγt+ innate lymphocytes and γδ T cells initiate psoriasiform plaque formation in mice. J. Clin. Investig. 2012, 122, 2252–2256.
- Soderstrom, C.; Berstein, G.; Zhang, W.; Valdez, H.; Fitz, L.; Kuhn, M.; Fraser, S. Ultra-sensitive measurement of IL-17A and IL-17F in psoriasis patient serum and skin. AAPS J. 2017, 19, 1218–1222.
- Ramirez-Carrozzi, V.; Sambandam, A.; Luis, E.; Lin, Z.; Jeet, S.; Lesch, J.; Hackney, J.; Kim, J.; Zhou, M.; Lai, J.; et al. IL-17C regulates the innate immune function of epithelial cells in an autocrine manner. Nat. Immunol. 2011, 12, 1159–1166.
- Chang, S.H.; Reynolds, J.M.; Pappu, B.P.; Chen, G.; Martinez, G.J.; Dong, C. Interleukin-17C promotes Th17 cell responses and autoimmune disease via interleukin-17 receptor E. Immunity 2011, 35, 611–621.
- Song, X.; Zhu, S.; Shi, P.; Liu, Y.; Shi, Y.; Levin, S.D.; Qian, Y. IL-17RE is the functional receptor for IL-17C and mediates mucosal immunity to infection with intestinal pathogens. Nat. Immunol. 2011, 12, 1151–1158.
- Song, X.; Gao, H.; Qian, Y. Th17 differentiation and their pro-inflammation function. Adv. Exp. Med. 2014, 841, 99–151.
- Reszke, R.; Szepietowski, J.C. Secukinumab in the treatment of psoriasis: An update. Immunotherapy 2017, 9, 229–238.
- Deodhar, A.; Mease, P.J.; McInnes, I.B.; Baraliakos, X.; Reich, K.; Blauvelt, A.; Leonardi, C.; Porter, B.; Gupta, A.D.; Widmer, A.; et al. Long-term safety of secukinumab in patients with moderate-to-severe plaque psoriasis, psoriatic arthritis, and ankylosing spondylitis: Integrated pooled clinical trial and post-marketing surveillance data. Arthritis Res. Ther. 2019, 21, 111.
- Yang, E.J.; Beck, K.M.; Liao, W. Secukinumab in the treatment of psoriasis: Patient selection and perspectives. Psoriasis (Auckl.) 2018, 8, 75.
- Kolbinger, F.; Loesche, C.; Valentin, M.A.; Jiang, X.; Cheng, Y.; Jarvis, P.; Peters, T.; Calonder, C.; Bruin, G.; Polus, F.; et al. β-Defensin 2 is a responsive biomarker of IL-17A–driven skin pathology in patients with psoriasis. J. Allergy Clin. Immunol. 2017, 139, 923–932.
- Toussirot, E. Ixekizumab: An anti-IL-17A monoclonal antibody for the treatment of psoriatic arthritis. Expert Opin. Biol. Ther. 2018, 18, 101–107.
- Blegvad, C.; Skov, L.; Zachariae, C. Ixekizumab for the treatment of psoriasis: An update on new data since first approval. Expert Rev. Clin. Immunol. 2019, 15, 111–121.
- Zaba, L.C.; Suárez-Fariñas, M.; Fuentes-Duculan, J.; Nograles, K.E.; Guttman-Yassky, E.; Cardinale, I.; Lowes, M.A.; Krueger, J.G. Effective treatment of psoriasis with etanercept is linked to suppression of IL-17 signaling, not immediate response TNF genes. J. Allergy Clin. Immunol. 2009, 124, 1022–1030.
- Blauvelt, A.; Reich, K.; Tsai, T.F.; Tyring, S.; Vanaclocha, F.; Kingo, K.; Ziv, M.; Pinter, A.; Vender, R.; Hugot, S.; et al. Secukinumab is superior to ustekinumab in clearing skin of subjects with moderate-to-severe plaque psoriasis up to 1 year: Results from the CLEAR study. JAAD 2017, 76, 60–69.
- Puig, L. Brodalumab: The first anti-IL-17 receptor agent for psoriasis. Drugs Today 2017, 53, 283–297.
- Foulkes, A.C.; Warren, R.B. Brodalumab in psoriasis: Evidence to date and clinical potential. Drugs Context 2019, 8, 212570.
- Russell, C.B.; Rand, H.; Bigler, J.; Kerkof, K.; Timour, M.; Bautista, E.; Krueger, J.G.; Salinger, D.H.; Welcher, A.A.; Martin, D.A. Gene expression profiles normalized in psoriatic skin by treatment with brodalumab, a human anti–IL-17 receptor monoclonal antibody. J. Immunol. 2014, 192, 3828–3836.
- Nirula, A.; Nilsen, J.; Klekotka, P.; Kricorian, G.; Erondu, N.; Towne, J.E.; Russell, C.B.; Martin, D.A.; Budelsky, A.L. Effect of IL-17 receptor A blockade with brodalumab in inflammatory diseases. Rheumatology 2016, 55, 43–55.
- Ridker, P.M.; Everett, B.M.; Thuren, T.; MacFadyen, J.G.; Chang, W.H.; Ballantyne, C.; Fonseca, F.; Nicolau, J.; Koenig, W.; Anker, S.D.; et al. Antiinflammatory therapy with canakinumab for atherosclerotic disease. NEJM 2017, 377, 1119–1131.
- Chiricozzi, A.; Saraceno, R.; Chimenti, M.S.; Guttman-Yassky, E.; Krueger, J.G. Role of IL-23 in the pathogenesis of psoriasis: A novel potential therapeutic target? Expert Opin. Ther. Targets 2014, 18, 513–525.
- Chan, T.C.; Hawkes, J.E.; Krueger, J.G. Interleukin 23 in the skin: Role in psoriasis pathogenesis and selective interleukin 23 blockade as treatment. Ther. Adv. Chronic Dis. 2018, 9, 111–119.
- Chan, J.R.; Blumenschein, W.; Murphy, E.; Diveu, C.; Wiekowski, M.; Abbondanzo, S.; Bowman, E.P. IL-23 stimulates epidermal hyperplasia via TNF and IL-20R2–dependent mechanisms with implications for psoriasis pathogenesis. Exp. Med. 2006, 203, 2577–2587.
- Chen, L.; Deshpande, M.; Grisotto, M.; Smaldini, P.; Garcia, R.; He, Z.; Gulko, P.S.; Lira, S.A.; Furtado, G.C. Skin expression of IL-23 drives the development of psoriasis and psoriatic arthritis in mice. Sci. Rep. 2020, 10, 8259.
- Di Meglio, P.; Nestle, F.O. The role of IL-23 in the immunopathogenesis of psoriasis. F1000 Biol. 2010, 2, 40.
- Fotiadou, C.; Lazaridou, E.; Sotiriou, E.; Ioannides, D. Targeting IL-23 in psoriasis: Current perspectives. Psoriasis: Targets Ther. 2018, 8, 1–5.
- Papp, K.; Thaçi, D.; Reich, K.; Riedl, E.; Langley, R.G.; Krueger, J.G.; Gottlieb, A.B.; Nakagawa, H.; Bowman, E.P.; Mehta, A.; et al. Tildrakizumab (MK-3222), an anti-interleukin-23p19 monoclonal antibody, improves psoriasis in a phase IIb randomized placebo-controlled trial. Br. J. Dermatol. 2015, 173, 930–939.
- Banaszczyk, K. Tildrakizumab in the treatment of psoriasis–literature review. Reumatologia 2019, 57, 234.
- Reich, K.; Papp, K.A.; Blauvelt, A.; Tyring, S.K.; Sinclair, R.; Thaçi, D.; Nograles, K.; Mehta, A.; Cichanowitz, N.; Li, Q.; et al. Tildrakizumab versus placebo or etanercept for chronic plaque psoriasis (reSURFACE 1 and reSURFACE 2): Results from two randomised controlled, phase 3 trials. Lancet 2017, 390, 276–288.
- Gordon, K.B.; Duffin, K.C.; Bissonnette, R.; Prinz, J.C.; Wasfi, Y.; Li, S.; Shen, Y.K.; Szapary, P.; Randazzo, B.; Reich, K. A phase 2 trial of guselkumab versus adalimumab for plaque psoriasis. NEJM 2015, 373, 136–144.
- Sweet, K.; Song, Q.; Loza, M.J.; McInnes, I.B.; Ma, K.; Leander, K.; Franks, C.; Cooper, P.; Siebert, S. Guselkumab induces robust reduction in acute phase proteins and type 17 effector cytokines in active psoriatic arthritis: Results from phase 3 trials. RMD Open 2021, 7, e001679.
- Jones, L.L.; Vignali, D.A. Molecular interactions within the IL-6/IL-12 cytokine/receptor superfamily. Immunol. Res. 2011, 51, 5–14.
- Lee, E.; Trepicchio, W.L.; Oestreicher, J.L.; Pittman, D.; Wang, F.; Chamian, F.; Dhodapkar, M.; Krueger, J.G. Increased expression of interleukin 23 p19 and p40 in lesional skin of patients with psoriasis vulgaris. J. Exp. Med. 2004, 199, 125–130.
- Jeon, C.; Sekhon, S.; Yan, D.; Afifi, L.; Nakamura, M.; Bhutani, T. Monoclonal antibodies inhibiting IL-12, -23, and-17 for the treatment of psoriasis. Hum. Vaccines Immunother. 2017, 13, 2247–2259.
- Smeltz, R.B.; Chen, J.; Ehrhardt, R.; Shevach, E.M. Role of IFN-γ in Th1 differentiation: IFN-γ regulates IL-18Rα expression by preventing the negative effects of IL-4 and by inducing/maintaining IL-12 receptor β2 expression. J. Immunol. 2002, 168, 6165–6172.
- Wilson, N.J.; Boniface, K.; Chan, J.R.; McKenzie, B.S.; Blumenschein, W.M.; Mattson, J.D.; Basham, B.; Smith, K.; Chen, T.; Morel, F.; et al. Development, cytokine profile and function of human interleukin 17–producing helper T cells. Nat. Immunol. 2007, 8, 950–957.
- El-behi, M.; Ciric, B.; Yu, S.; Zhang, G.X.; Fitzgerald, D.C.; Rostami, A. Differential effect of IL-27 on developing versus committed Th17 cells. J. Immunol. 2009, 183, 4957–4967.
- Parham, C.; Chirica, M.; Timans, J.; Vaisberg, E.; Travis, M.; Cheung, J.; Pflanz, S.; Zhang, R.; Singh, K.P.; Vega, F.; et al. A receptor for the heterodimeric cytokine IL-23 is composed of IL-12Rβ1 and a novel cytokine receptor subunit, IL-23R. J. Immunol. 2002, 168, 5699–5708.
- Zaghi, D.; Krueger, G.G. Ustekinumab: A review in the treatment of plaque psoriasis and psoriatic arthritis. J. Drugs Dermatol. 2012, 11, 160–167.
- Brodmerkel, C.; Li, K.; Garcet, S.; Hayden, K.; Chiricozzi, A.; Novitskaya, I.; Krueger, J.G. Modulation of inflammatory gene transcripts in psoriasis vulgaris: Differences between ustekinumab and etanercept. J. Allergy Clin. Immunol. 2019, 143, 1965–1969.
- Tohyama, M.; Yang, L.; Hanakawa, Y.; Dai, X.; Shirakata, Y. IFN-α enhances IL-22 receptor expression in keratinocytes: A possible role in the development of psoriasis. J. Investig. Dermatol. 2012, 2132, 1933–1937.