The donkey hindgut is a microbial-rich ecosystem in which caecum and colon fungi play an important role in dietary fiber degradation. In addition, the fibrolytic enzymes produced by hindgut microorganisms are key to the ability of equines to hydrolysis plant fiber. In the entry, the fibrolytic enzyme activities within donkey caecum and colon were firstly measured by spectrophotometry. The dorsal colon presented a higher fibrolytic enzyme activity in comparison with caecum. The fungal community composition along donkey caecum and colon was determined by sequencing an internal transcribed spacer region (ITS) using Illumina MiSeq. The predominant fungi at phylum level were Ascomycota, Basidiomycota, and Neocallimastigomycota. The Aspergillus, Wallemia, Phanerochaete, Fusarium, and Penicillium were detected as the dominant genera, but their metabolic and functional significance in donkey cecum-colon ecosystem need further investigation. In terms of the anaerobic fungi Neocallimastigomycota, its abundance was greater in donkey colon than in caecum. The relative abundance of enzymes related to plant cell wall breakdown were also predicted by PICRUSt, and they were also greater in donkey colon than in caecum. The entry provided new information about fibrolytic enzyme profiles and fungal communities in donkey hindgut. The findings could therefore contribute to the further understanding of the fungal taxa and their dietary fiber degradation mechanisms in donkey hindgut ecosystem.
1. Introduction
Donkeys have evolved as free-grazing herbivores with a specialized and enlarged hindgut. The equine hindgut is mainly comprised of two fermentative compartments, the caecum and colon, which together account for two-thirds of the volume of donkey’s gastrointestinal tract
[1]. Microbial fermentation of plant structural polysaccharides in donkey hindgut results in the production of volatile fatty acids (VFAs), which are a major source of energy for the host
[2].
Among hindgut microorganisms, both the bacteria and the fungi play a pivotal role in the hindgut fermentation and the degradation of feedstuffs
[2]. Nowadays, increasing studies have been successfully performed to reveal the changes and functions of the hindgut bacteria community in equines
[3][4]. However, little information is available regarding the composition of fungal communities in the equine hindgut ecosystem. The current knowledge of gut fungi has focused on the strictly anaerobic fungi (phylum
Neocallimastigomycota) and is mostly derived from ruminant based studies
[5][6].
Based on physiological and developmental characteristics,
Neocallimastigomycota is generally divided into the monocentric rhizoidal, the polycentric rhizoidal, and the bulbous genera
[5]. Anaerobic fungi were reported to be present in domesticated and non-domesticated equine species, and their diversity in donkeys was shown to be higher when compared to that of ponies and pony × donkey hybrids
[2]. Recently, Edwards et al. determined the fecal community composition of anaerobic fungi in donkeys, and the genus
Caecomyces, SK3, KF1, and
Piromyces were found in the fecal samples
[2]. The presence of these anaerobic fungi may enable the donkeys to utilize fibrous feeds efficiently and persistently. However, recent studies in ruminant animals have shown that
Ascomycota and
Basidiomycota phyla also play an important role in the rumen digestion of cashmere goats and Holstein cows
[7][8].
The fungal ribosomal RNA (rRNA) operon-based analysis has facilitated the understanding of the diversity and phylogenetic relationships of gut fungi in natural ecosystems
[9]. The internal transcribed spacer (ITS) region is usually recommended as the DNA barcode marker for gut fungi
[10][11]. In addition, the 28S ribosomal large subunit (28S rRNA gene (LSU); D1/D2 region) might render higher coverage of early diverging lineages of fungi, such as
Neocallimastigomycota,
Chytridiomycota, and
Mucoromycota [11]. However, there is still a big challenge of how to relate LSU taxonomic sequences to the clades that until now only contained “unculturable representatives” derived ITS1 sequences
[5][12]. Therefore, the ITS region remains the most accepted phylogenetic marker to assess the diversity and community structure of gut fungi.
Due to ethical noninvasive sampling, fecal samples are commonly used for fungi investigation in equines
[2][7]. The understanding of equine fungi diversity and community structure within different hindgut regions is limited. It is very necessary to evaluate the diversity and composition of hindgut fungi in the anatomically specialized hindgut of donkeys. Moreover, both the bacteria and the fungi produce a broad array of fibrolytic enzymes that can facilitate the plant fragments degradation by breaking the linkages between lignin and hemicelluloses
[13][14]. The digestibility of plant fiber in donkeys may be to a large extent dependent on the strong fibrolytic activity of the main-chain or the side-chain degrading enzymes, including carboxymethyl cellulase, xylanase, ferulic acid esterase, and acetyl esterase. However, neither the main-chain degrading enzymes nor the side-chain degrading enzymes have been reported in the donkey caecum and colon.
2. Fibrolytic Enzyme Profiles
Apart from the FAE, the activities of CMCase, AVI, XLY, and AE in caecum and ventral colon were all lower than that in dorsal colon (
Table 1,
p < 0.05).
Table 1. The fibrolytic enzyme profiles in cecum, ventral colon, and dorsal colon of Dezhou donkeys.
| Items |
Caecum |
Ventral Colon |
Dorsal Colon |
SEM |
p |
| CMCase, U |
0.13 b |
0.10 b |
0.37 a |
0.066 |
0.02 |
| AVI, U |
0.14 b |
0.12 b |
0.23 a |
0.015 |
<0.01 |
| XLY, U |
2.5 b |
2.0 b |
7.3 a |
0.570 |
<0.01 |
| FAE, mU |
4.2 |
4.5 |
4.5 |
0.47 |
0.85 |
| AE, mU |
21.6 b |
20.2 b |
30.2 a |
1.72 |
<0.01 |
a,b Values in the same row with different small letter superscripts represent significant difference (p < 0.05), while with no letter or the same letter superscripts represent no significant difference (p > 0.05). CMCase, carboxymethyl cellulase; AVI, avicelase; XLY, xylanase; FAE, ferulic acid esterase; AE, acetyl esterase; SEM, mean standard error.
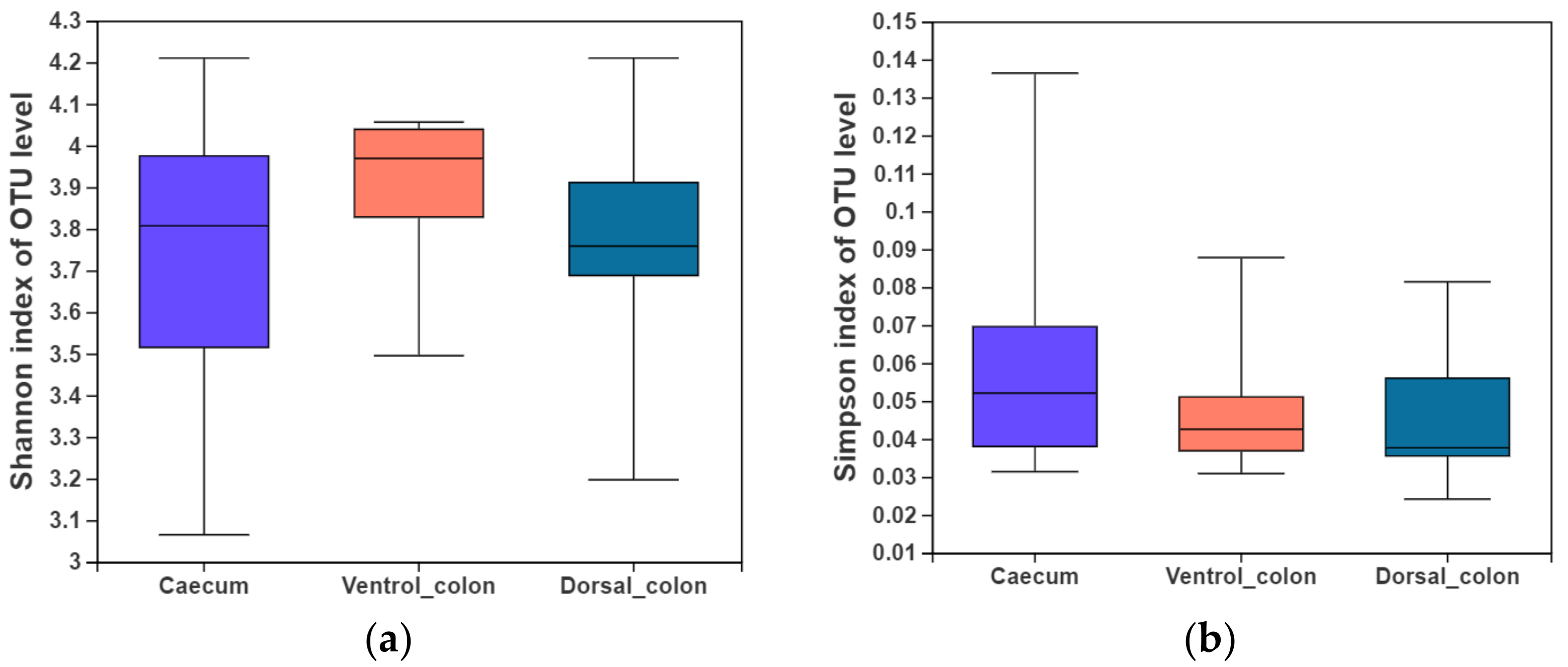
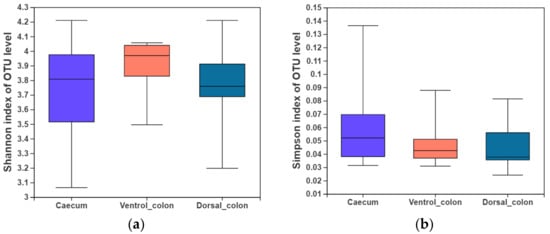
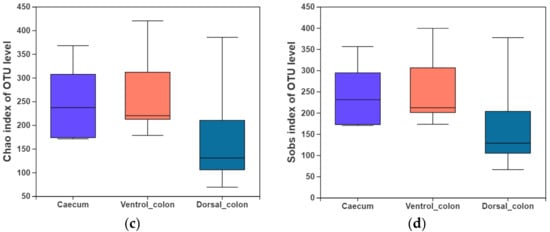
3. Alpha Diversity
In the present study, indices of alpha diversity included Shannon, Simpson, Chao, and Sobs index (
Figure 1). No difference occurred among donkey caecum, ventral colon, and dorsal colon (
p > 0.05).
Figure 1. Fungal alpha diversity indices of caecum and colon in Dezhou donkeys. (a), Shannon index; (b), Simpson index; (c), Chao index; (d), Sobs index.
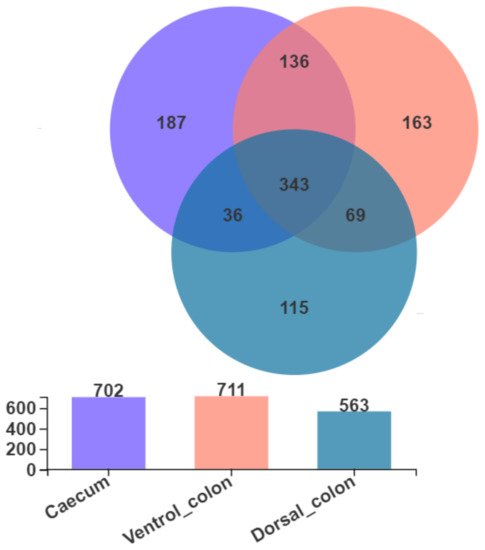
4. Fungal Community Composition
As shown in
Figure 2, Venn diagram presented the distribution of fungal community OTUs. There were 702, 711, and 563 OTUs that were observed in donkey caecum, ventral colon, and dorsal colon, respectively. In addition, caecum shares the fungal community, including 343 OTUs, with ventral colon and dorsal colon.
Figure 2. Venn diagram presented the distribution of fungal community OTUs across the caecum, ventral colon, and dorsal colon.
Fungi with a relative abundance of ≥1% of the total sequences in at least one of the samples were further analyzed (
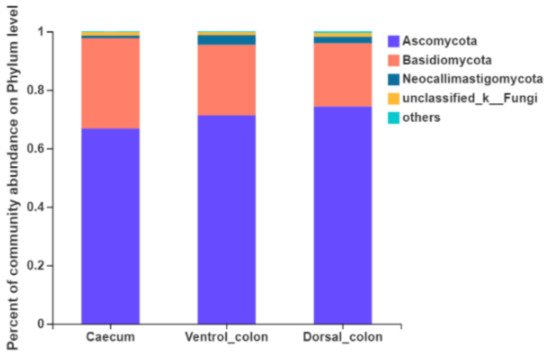
Figure 3). The top four predominant phylum were
Ascomycota (66.8%–74.4% of the total sequence reads),
Basidiomycota (21.6%–30.9%),
Neocallimastigomycota (0.9%–3.3%) and unclassified fungi (1.0%–1.3%).
Figure 3. Composition of the predominant fungal phyla (relative abundance ≥1%) among donkey caecum, ventral colon, and dorsal colon (n = 8).
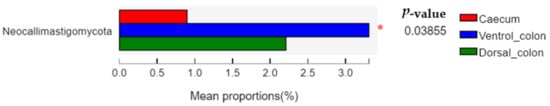
The relative abundance of
Neocallimastigomycota in donkey caecum was significantly lower than that in the ventral and dorsal colon (
Figure 4;
p = 0.03).
Figure 4. Difference in the relative abundance of fungal phyla (abundance of the phyla was expressed as %). Welch’s two-sided test was used and Welch’s inverted was 0.95. *, p < 0.05.
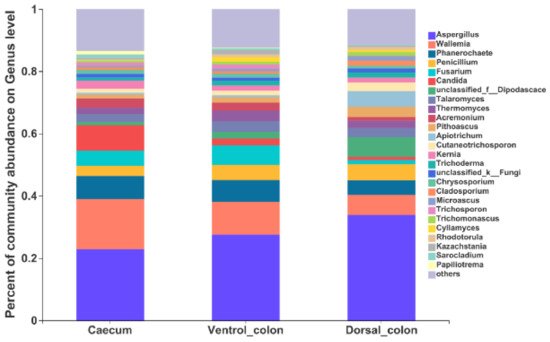
At the genus level, the top 10 predominant genera were
Aspergillus, Wallemia, Phanerochaete, Fusarium, Candida, unclassified_
Dipodascace, Talaromyces, Thermomyces, Acremonlum, and
Pithoascus (
Figure 5).
Figure 5. Composition of the fungal genera (relative abundance >1%) among donkey caecum, ventral colon, and dorsal colon (n = 8).
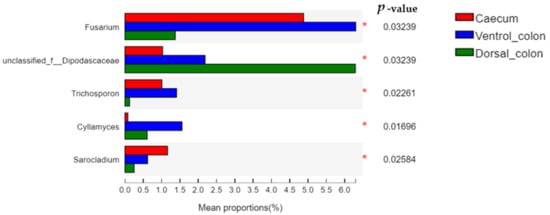
The relative abundance of
Fusarium and
Trichosporon in donkey hindgut was ranked as: ventral colon > caecum > dorsal colon (
Figure 6;
p < 0.05). In addition, the relative abundance of unclassified
Dipodascaceae and
Cyllamyces in donkey colon was significantly greater than that in the caecum (
p < 0.05).
Figure 6. Difference in the relative abundance of fungal genera (abundance of the phyla was expressed as %). Welch’s two-sided test was used and Welch’s inverted was 0.95. *, p < 0.05.
The anaerobic fungal community belonging to the
Neocallimastigomycota phylum were further analyzed (
Table 2). The genera
Cyllamyces, Neocallimastix, Piromyces, Buwchfawromyces, Unclassified_f_
Neocallimastigaceae, and unclassified_c_
Neocallimastigomycetes were detected within donkey hindgut. The abundance of anaerobic fungal community composition varied greatly between donkey individuals. In addition, the relative abundance of anaerobic fungi was numerically lower in the caecum than in both ventral colon and dorsal colon.
Table 2. The relative abundance of anaerobic fungal genera composition (belonging to the Neocallimastigomycota phylum) in donkey caecum and colon.
| Items |
|
Caecum |
Ventral Colon |
Dorsal Colon |
| Cyllamyces |
Min |
0.008 |
0.011 |
0.002 |
| Max |
0.589 |
3.81 |
2.16 |
| Mean ± SD |
0.21 ± 0.19 |
0.71 ± 0.52 |
0.69 ± 0.32 |
| Piromyces |
Min |
0.002 |
0.002 |
0.009 |
| Max |
0.346 |
1.21 |
3.34 |
| Mean ± SD |
0.12 ± 0.11 |
0.42 ± 0.18 |
1.24 ± 1.05 |
| Neocallimastix |
Min |
ND |
0.003 |
ND |
| Max |
0.002 |
2.67 |
5.66 |
| Mean ± SD |
0.002 |
0.68 ± 0.66 |
5.66 |
| Buwchfawromyces |
Min |
0.006 |
0.003 |
ND |
| Max |
0.014 |
1.88 |
0.95 |
| Mean ± SD |
0.01 ± 0.004 |
0.94 ± 0.93 |
0.95 |
| Unclassified_f_Neocallimastigaceae |
Min |
0.002 |
0.013 |
0.049 |
| Max |
0.066 |
4.78 |
0.838 |
| Mean ± SD |
0.03 ± 0.02 |
0.83 ± 0.57 |
0.41 ± 0.13 |
| Unclassified_c_Neocallimastigomycetes |
Min |
ND |
0.002 |
0.003 |
| Max |
ND |
0.18 |
0.03 |
| Mean ± SD |
ND |
0.06 ± 0.04 |
0.02 ± 0.01 |
Min, minimum value; Max, maximum value; SD, standard error; ND, not detected.
5. Beta Diversity
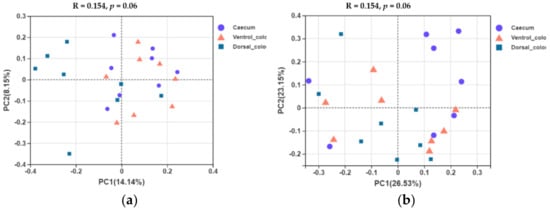
In terms of beta diversity at the OTU level, unweighted and weighted unifrac PCoA showed that there were obvious varieties between individuals for the fungal community composition. Separation of dorsal colon from caecum occurred along the first axis (PC1) of the unweighted PCoA, but no separation of caecum from ventral colon was seen (
Figure 7a). No obvious separation of the samples by hindgut region was observed in the weighted PCoA (
Figure 7b).
Figure 7. Unweighted (a) and weighted (b) unifrac based principal coordinates analysis (PCoA) of the fungal community composition of the different hindgut regions at the OTU level. Analysis used Log10 transformed data, and the percentage values given on each axis indicate the amount of total variation represented. PC1, first axis; PC2, second axis.
6. Prediction of Fungal Enzymatic Activity by PICRUSt (Phylogenetic Investigation of Communities by Reconstruction of Unobserved States)
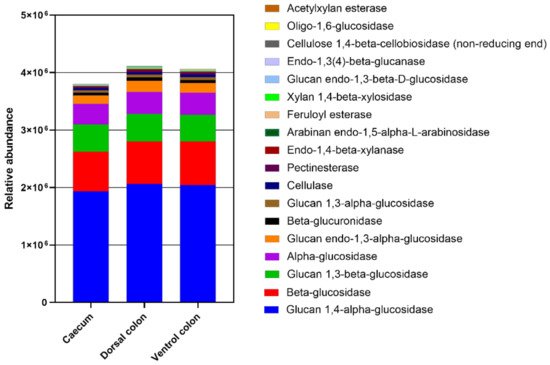
The relative abundance of enzymes related to plant cell wall degradation were predicted by PICRUSt (
Figure 8). These enzymes mainly included glucan 1,4-alpha-glucosidase beta-glucosidase, glucan 1,3-beta-glucosidase, alpha-glucosidase, glucan endo-1,3-alpha-glucosidase, beta-glucuronidase, glucan 1,3-alpha-glucosidase, cellulase, pectinesterase, endo-1,4-beta-xylanase, arabinan endo-1,5-alpha-L-arabinosidase, feruloyl esterase, xylan 1,4-beta-xylosidase, glucan endo-1,3-beta-D-glucosidase, endo-1,3(4)-beta-glucanase, cellulose 1,4-beta-cellobiosidase (non-reducing end), and oligo-1,6-glucosidase, acetylxylan esterase. The relative abundances of these enzymes were lower in caecum than in both ventral and dorsal colon.
Figure 8. The abundance of enzymes related to plant cell wall degradation predicted by phylogenetic investigation of communities by reconstruction of unobserved states (PICRUSt). PICRUSt is a bioinformatics software package designed to predict metagenome functional content from marker gene (e.g., ITS) surveys.