Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Camila Xu and Version 1 by María Luisa Ojeda Murillo.
Selenium (Se) (Se 34 79) is an essential trace element mainly known for its antioxidant [1], anti-inflammatory, and anti-apoptotic properties as it is part of the catalytic center of different selenoproteins with different enzymatic activities. During different pathologies Se tissue concentration and selenoproteins expression are modified, afecting tissue function.
- selenium
- selenoprotein
- metabolic syndrome
- oxidative balance
- insulin resistance
- thyroid function
- development
1. Selenium
Selenium (Se) (Se 34 79) is an essential trace element mainly known for its antioxidant [1], anti-inflammatory, and anti-apoptotic properties as it is part of the catalytic center of different selenoproteins [2,3,4][2][3][4]. Adequate Se intake is essential for immune, endocrine, cardiovascular, reproductive, and nervous systems functions [5,6,7,8,9][5][6][7][8][9]. However, the margin between Se toxicity and its deficiency is very narrow [10]. Its role in the human body has been studied, especially for thyroid function, type 2 diabetes mellitus (T2DM), hypertension, obesity, inflammation, reproductive system, cancer, and cardiovascular disease. Se excessive and deficient dietary intake is associated with damaging health effects that have been characterized by a U-shaped relationship [11]. For this, a balanced intake of Se is crucial to maximizing the health benefits of selenium [12]. Recommendations for Se intake are: in adults 55–70 µg/day, in infants 15 µg/day, in children 20–30 µg/day, in pregnant women 65 µg/day, and in lactating mothers 75 µg/day [13], the tolerable upper intake level is limited to 300 µg/day [14].
Dietary Se is absorbed by the gastrointestinal tract (GIT) in its inorganic and organic forms. Organic forms of Se such as selenomethionine (SeMet) and selenocysteine (Sec) are absorbed in the small intestine through the same active sodium-dependent transport system as the amino acid methionine. Additionally, Sec may be absorbed using the same mechanism as cysteine [15,16,17,18][15][16][17][18]. Inorganic forms are absorbed by the same sodium-facilitated and energy-dependent systems as sulfate [19]. Moreover, selenate and selenite can be uptaken by non-mediated passive diffusion, with a slower absorption rate than the Se-organic compounds [20]. After intestinal absorption, Se forms enter the bloodstream and are predominantly taken up into the liver from the portal vein [21]; in this tissue they will be further metabolized, turning these inorganic forms into more bioavailable organic forms. So, in the human body, two metabolic pools of Se are predominant. One pool includes all forms of Se derived from inorganic selenite/selenide, including excretory Se metabolites and other intermediate products of selenite metabolism [22]. The second pool consists of organic forms of Se and SeMet-containing proteins. The extracellular Se forms, from inorganic and organic pools, are captured by the liver and other tissues such as muscle and mammary glands [23]. Later inside the cells, Sec, selenite, and selenide compounds form an intracellular metabolic reserve, whereas SeMet is incorporated into proteins in place of methionine. This amino acid is also converted to Sec via the transsulfuration pathway, and this Sec is transformed to selenide with the help of Sec-lyase enzyme. In the liver, inorganic forms are reduced to selenide by thioredoxin reductases or the glutathione (GSH) pathway. Then selenide is transformed to selenophosphate by the enzyme selenophosphate synthetase 2. Selenophosphate is used for selenoprotein synthesis.
The selenoprotein synthesis (translational decoding process) begins when selenophospate reactions with phosphoseryl-tRNA yield Sec-tRNA[Ser]Sec. Sec amino acids are incorporated into polypeptide chains utilizing the UGA codon. Selenocysteine insertion sequence binding protein 2 (SBP2) binds to selenocysteine insertion sequence (SECIS) element which is located in the 3′-untranslated region (3′UTR) of selenoprotein mRNA and mediates the transfer of Sec-tRNA[Ser]Sec to the A-site of the ribosome which recognizes the UGA codon as the Sec integration codon [24]. The amino acid Sec can appear in the N or C-terminal part of protein according to two different groups of selenoproteins, representing the most important part of the active center of selenoproteins. Selenoprotein P (SelP) is the main selenoprotein produced in the liver, containing 10 Sec residues; so, it functions as a Se-transport protein to deliver Se to other tissues [25]. The different tissues need specific receptors to uptake SelP. Then, in the tissues, Se is used to synthetize new other selenoproteins [26,27,28][26][27][28]. The placenta, brain, and testes uptake SelP through receptor-mediated endocytosis using the low-density lipoprotein receptor-related protein 8 (LRP8), also known as ApoER2 [29,30][29][30]. Other tissues, like the kidneys, also use another membrane receptor, the megalin or LRP2. Se is differentially distributed in the body, following a tissue hierarchy that is even extended to intracellular mechanisms that prioritize the synthesis of specific selenoproteins [31,32,33][31][32][33]. This fact makes the studies that address the tissue distribution of Se and selenoproteins extremely complex.
Twenty-five selenoproteins are recognized in humans, with different biological functions (Table 1) [34[34][35],35], such as iodothyronine deiodinases (DIOs: 1, 2, and 3 families), responsible for thyroid function; or thioredoxin reductases (TXNRD1, 2, and 3), SelW, SelH, SelT, and SelV, involved in redox regulation processes [36]. However, most of them have antioxidant properties, like the glutathione peroxidases family (GPxs: GPx1–GPx8), which eliminate the excess of H2O2, and selenoprotein P (SelP), the main serum Se transporter [2]. Both of them are also related to the endocrine system and intracellular signaling, appetite, growth, and energy homeostasis [37,38][37][38].
Table 1. Main selenoproteins, their physiological functions, relation to Se status, and pathophysiological implications (modified from Qazi et al. [39] and Hariharan et al. [2]).
| Selenoproteins | Physiological Functions | Relation to Se Status and Pathophysiological Implications |
|---|---|---|
| GPx1 (cytoplasmatic) |
Antioxidant | Sensitive to Se intake. Cardiovascular diseases. Related to the endocrine system, intracellular signaling, appetite, growth, energy homeostasis, and IR. |
| GPx2 (gastrointestinal) |
Antioxidant | Resistant to Se modifications. Intestinal cancer. |
| GPx3 (plasmatic) |
Extracellular fluid antioxidant | Sensitive to Se intake. Cardiovascular protection. |
| GPx4 (membranes) |
Membrane antioxidant. In sperm is a structural protein. Apoptosis | Resistant to Se modifications. Immune disorders, HIV, implications in male fertility, and mitochondrial function. |
| GPx6 (olfactory) |
Homolog to GPx3 | The knowledge of this GPx is very limited. |
| DIO1 | Conversion of T4 to T3 | Implications in immune thyroid disease and thyroid dysfunctions. |
| DIO2 | Conversion of T4 to T3 | Stable expression under low Se levels. Implications in immune thyroid disease and thyroid dysfunctions. |
| DIO3 | Conversion of T4 to reverseT3 | Implications in immune thyroid disease and thyroid dysfunctions. |
| TXNRD1 | Antioxidant, redox regulation, cell signaling | Se dependent. In several types of cancer, there is an over-expression of TXNRD1. |
| TXNRD2 | Antioxidant, redox regulation, cell signaling | Sensitive to Se intake. |
| TXNRD3 | Antioxidant, redox regulation, cell signaling | Role in sperm maturation. |
| SelW | Antioxidant | Studies with tissue cultures of muscle and brain cells indicated that Se influenced SelW levels. |
| SelH | GSH synthesis | Implications in placenta oxidative stress. |
| SelV | Redox regulation processes | Although it is expressed in seminiferous tubules in mice, the exact role in spermatogenesis is unknown. |
| SelT | Endoplasmic reticulum homeostasis: promotes depletion of Ca stores and impaired hormone secretion | Unknow, although probably related with Endoplasmic Reticulum Stress. |
| SelP (plasma) |
Main plasma Se transporter. Antioxidant. Indicator of Se status | Cancer, neurodegenerative diseases. Implicated in male fertility and maternal-fetal Se transfer. Apoptosis regulation. Related to the endocrine system, intracellular signaling, appetite, growth, energy, and IR. |
Among all these selenoproteins, the TXNRD, GPx, and DIO families are the three best characterized [40]. They have different enzymes activities, although all of them require reductants to provide the electrons to make their catalytic redox cycle run. Thus, TXNRD-dependent reduction requires electron transfer from NADPH, FAD via, to Sec at their active site and finally to the substrate thioredoxin. In the GPx family, the catalytic redox cycle involves the oxidation of Sec to selenic acid by hydrogen peroxide and organic hydroperoxides, and reduction to the selenolate anion form by the GSH system [41,42][41][42]. Finally, the action mechanism of DIO selenoproteins involves the generation of an oxidized intermediate, which will be reduced by thiol-containing reductants and release iodide.
In general terms, selenoproteins and Se can exert their main functions through different mechanisms. (1) By ROS-mediated stimulation of intracellular protein kinases in the cytoplasm and the nucleus, such as the mitogen-activated (MAP) kinase, the p38 kinase, and the c-jun/stress-activated kinase, all of them are involved in the growth responses of cells to stressful and inflammatory stimuli [43]. In this contHerext, several studiresearches have found that selenate is a stimulator of the tyrosine kinases, as happens in the insulin signaling cascade having insulin-mimetic effects [44[44][45][46],45,46], by contrast, high doses of selenite has shown to impair/dampen insulin signaling [47,48][47][48]. Therefore, Se compounds and selenoproteins play an important role in fuel metabolism processes [49,50][49][50]. (2) Another mechanism involves ROS-mediated covalent modification of thiol, cysteine, and tyrosine groups of proteins. (3) Additionally, Se can produce alterations in cellular redox state causing activation of transcription factors, such as NF-kB, Ap-1, and the glucorticoid receptor, leading to de novo gene expression. Thus, Se can affect transcription factors activation by either affecting DNA-binding strength or changing activation of the transcription factor by modulation of regulatory subunits, e.g., by phosphorylation. (4) Se regulates the expression of cell surface and nuclear receptors leading to alteration in cell growth, responsiveness, and behavior. (5) Finally, this element can regulate the cell death/survival signals.
Therefore, selenoproteins have an important role in insulin resistance (IR) and metabolic syndrome (MetS) generation, modulating the reactive oxygen species (ROS) implicated in insulin signaling and the energetic sensor AMP-activated protein kinase (AMPK) [46,51][46][51]. Elevated dietary Se intake is associated with IR by increasing hepatic GPx1 activity, which decreases H2O2 levels (Figure 1). In this case, H2O2 acts as a second messenger formed after the activation of NADPH oxidases (NOX) when insulin binds to its receptor (IR). H2O2 is required to inactivate various insulin-signaling inhibitors as the protein tyrosine phosphatase 1B (PTP-1B) or the dual-specificity phosphatase PTEN by oxidation of essential SH groups [52]. PTP-1B inhibits the phosphorylation (activation) of insulin receptor substrate (IRS-1), while PTEN dephosphorylates phosphatidylinositol 3,4,5-trisphosphate (PIP3) at position 3′ to PIP2, depressing protein kinase B (Akt) signaling and triggering IR [53]. Thus, high Se levels augment hepatic GPx1 activity, which reduces H2O2, decreasing the oxidative inhibition of PTP1B and PTEN and suppressing insulin signaling [54]. On the other hand, when SelP is increased in the liver it inhibits AMPK activity impairing insulin signaling transduction, and is recognized as a hepatokine that contributes to the beginning of hyperglycemia and IR [51]. Additionally, other mechanisms have been proposed to explain the role of SelP in IR using primary hepatocytes, describing that purified SelP induces a reduction in insulin-stimulated phosphorylation of IR, IRS-1, and Akt [55]. In addition, it is important to take in mind that supranutritional Se modifies the expression of transcriptional factors and enzymes related to carbohydrate, lipids, and protein metabolism in a tissue-selective manner, such as liver, skeletal muscle, and adipose tissue [50].
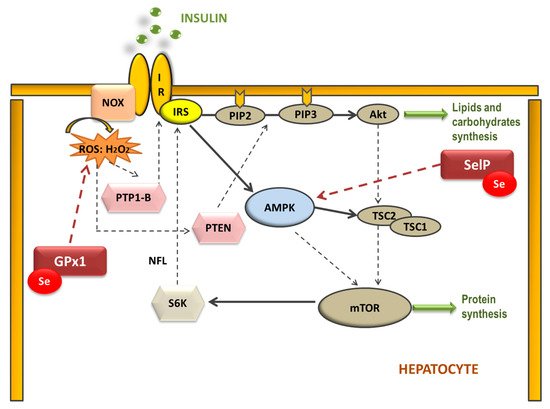
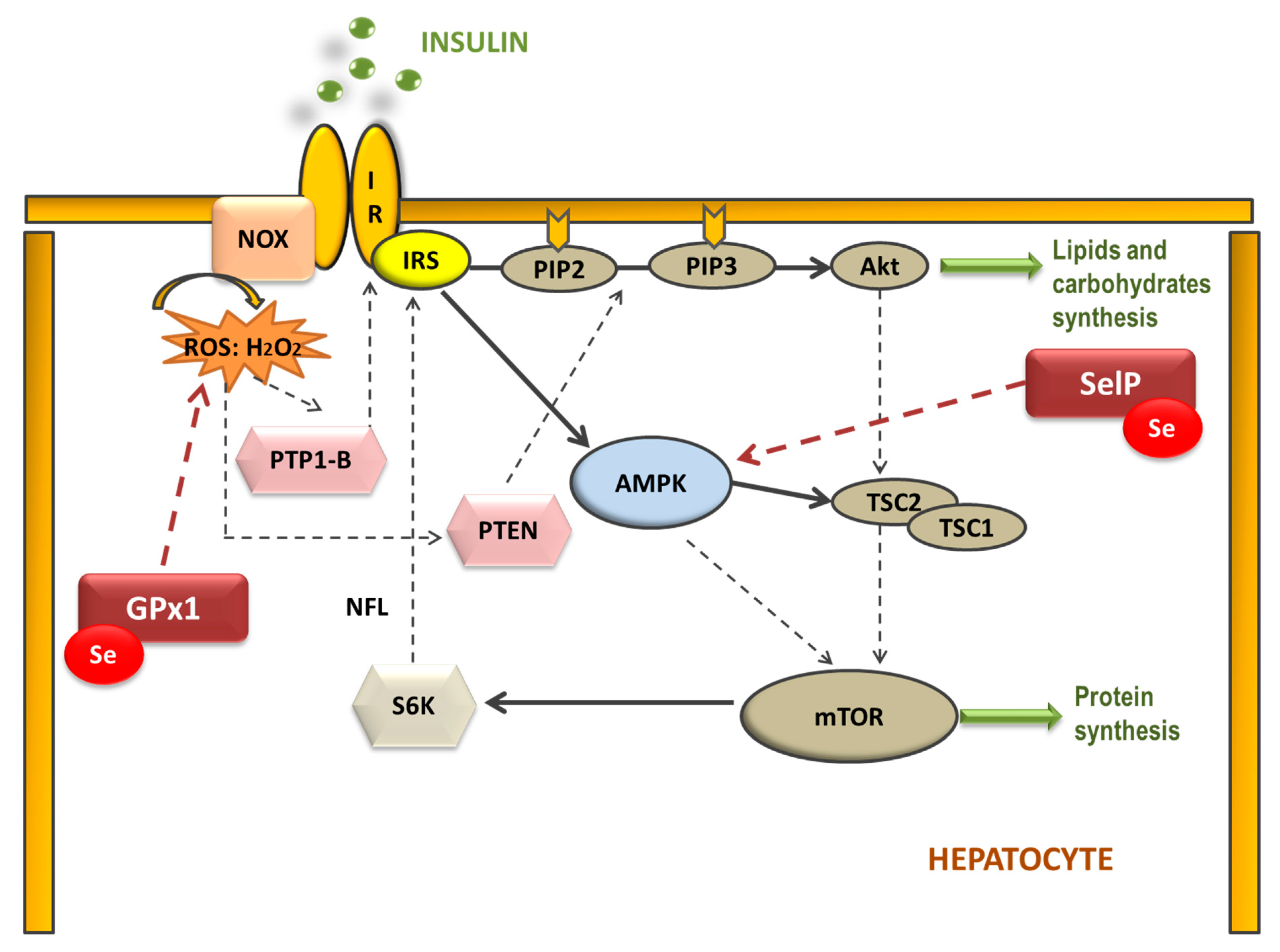 Figure 1. Selenoproteins’ possible implication in the hepatic insulin signaling pathway. The binding of insulin to the insulin receptor (IR) triggers consecutive phosphorylation (P) of downstream signaling molecules, resulting in activation of Akt. Additionally, NADPH oxidase (NOX)-mediated ROS production is stimulated by insulin. Reduction of H2O2 by GPx1 may attenuate insulin signaling, as H2O2 is required to inactivate the insulin counter-regulatory phosphatases PTP-1B and PTEN. Moreover, SelP could inhibit adenosine monophosphate-activated protein kinase (AMPK), a metabolic energy sensor, which negatively regulates protein synthesis through the inhibition of the mammalian target of rapamycin (mTOR)-S6 kinase (S6K) pathway, which also modulates IRS phosphorylation. (IRS: insulin receptor substrate; PIP2: phosphatidylinositol 4,5-bisphosphate; PIP3: phosphatidylinositol 3,4,5-trisphosphate; TSC1 and 2: tuberous sclerosis complex 1 and 2).
Figure 1. Selenoproteins’ possible implication in the hepatic insulin signaling pathway. The binding of insulin to the insulin receptor (IR) triggers consecutive phosphorylation (P) of downstream signaling molecules, resulting in activation of Akt. Additionally, NADPH oxidase (NOX)-mediated ROS production is stimulated by insulin. Reduction of H2O2 by GPx1 may attenuate insulin signaling, as H2O2 is required to inactivate the insulin counter-regulatory phosphatases PTP-1B and PTEN. Moreover, SelP could inhibit adenosine monophosphate-activated protein kinase (AMPK), a metabolic energy sensor, which negatively regulates protein synthesis through the inhibition of the mammalian target of rapamycin (mTOR)-S6 kinase (S6K) pathway, which also modulates IRS phosphorylation. (IRS: insulin receptor substrate; PIP2: phosphatidylinositol 4,5-bisphosphate; PIP3: phosphatidylinositol 3,4,5-trisphosphate; TSC1 and 2: tuberous sclerosis complex 1 and 2).However, Se deficiency is also associated with IR [56,57][56][57]. Seale et al. found that when cellular Se recycling mechanisms are lacking, a decrease in GPx1 and SelP expression appears, which is related to an inhibition of insulin signaling; probably, among others, by leading to an excessive increase of H2O2 and oxidative stress (OS) [58]. These results have been recently confirmed by different authors, who concluded that a disruption on the selenocysteine lyase which mediates the Se recycling pathway, leads to hyper-adiposity, obesity, IR, and deep changes in metabolic homeostasis [59,60,61][59][60][61]. For these reasons, an appropriate Se homeostasis, neither high nor low, is important to maintain a correct oxidative balance in order to avoid metabolic disruptions.
OS is a state of negative imbalance between the excess of pro-oxidative compounds and the insufficient decomposition of those compounds by the antioxidant systems [62]. ROS in basal conditions are essential for cellular functions (such as in the insulin signaling cascade), whereas excessive levels of ROS cause damage to cells by the oxidation of lipids, DNA, and proteins [63[63][64][65],64,65], leading to cellular dysfunction including loss of energy metabolism, altered cell signaling and cell cycle control, genetic mutations, altered cellular transport mechanisms, and overall decreased biological activity, immune activation, and inflammation [66]. ROS include oxygen free radicals, such as the superoxide anion (O2−), the hydroxyl radical (•OH), and non-radicals, such as hydrogen peroxide (H2O2). They can also react with NO leading to peroxynitrite, the so-called RNS. The defense mechanism responsible for ROS inactivation included endogenous antioxidants, classified into enzymatic antioxidants (GPxs, superoxide dismutase (SOD) and catalase (CAT)), and non-enzymatic antioxidants (nicotinamide adenine dinucleotide) and reduced glutathione (GSH), and exogenous antioxidants, such as some vitamins and metals [67]. The SOD enzyme constitutes the first line of defense against free radicals by catalyzing the dismutation of O2− to H2O2 and oxygen decreasing O2− concentration, which damages the cells at an excessive concentration [68]. H2O2 formed is not a radical, but it is rapidly converted by the Fenton reaction into •OH radical which is very reactive. For this reason, there are two antioxidant enzymes, catalase and the selenoprotein GPx, responsible for reducing it. GPx neutralizes H2O2 by taking hydrogens from two GSH molecules resulting in two H2O and one oxidized glutathione (GSSG). GR, an NADPH-dependent enzyme, is fundamental in this step since it regenerates GSH from GSSG. The GPx enzyme is more important against high H2O2 levels; however, catalase acts preferentially when there are low concentrations of H2O2. Besides, GPx is also responsible for detoxifying other lipid peroxides (LOOH) to the corresponding alcohol (LOH). The defense mechanism of endogenous antioxidant enzymes is shown in Figure 2.
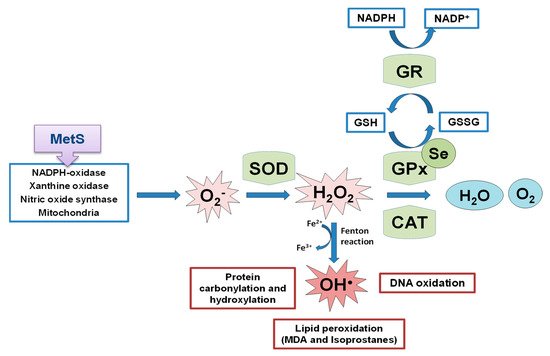
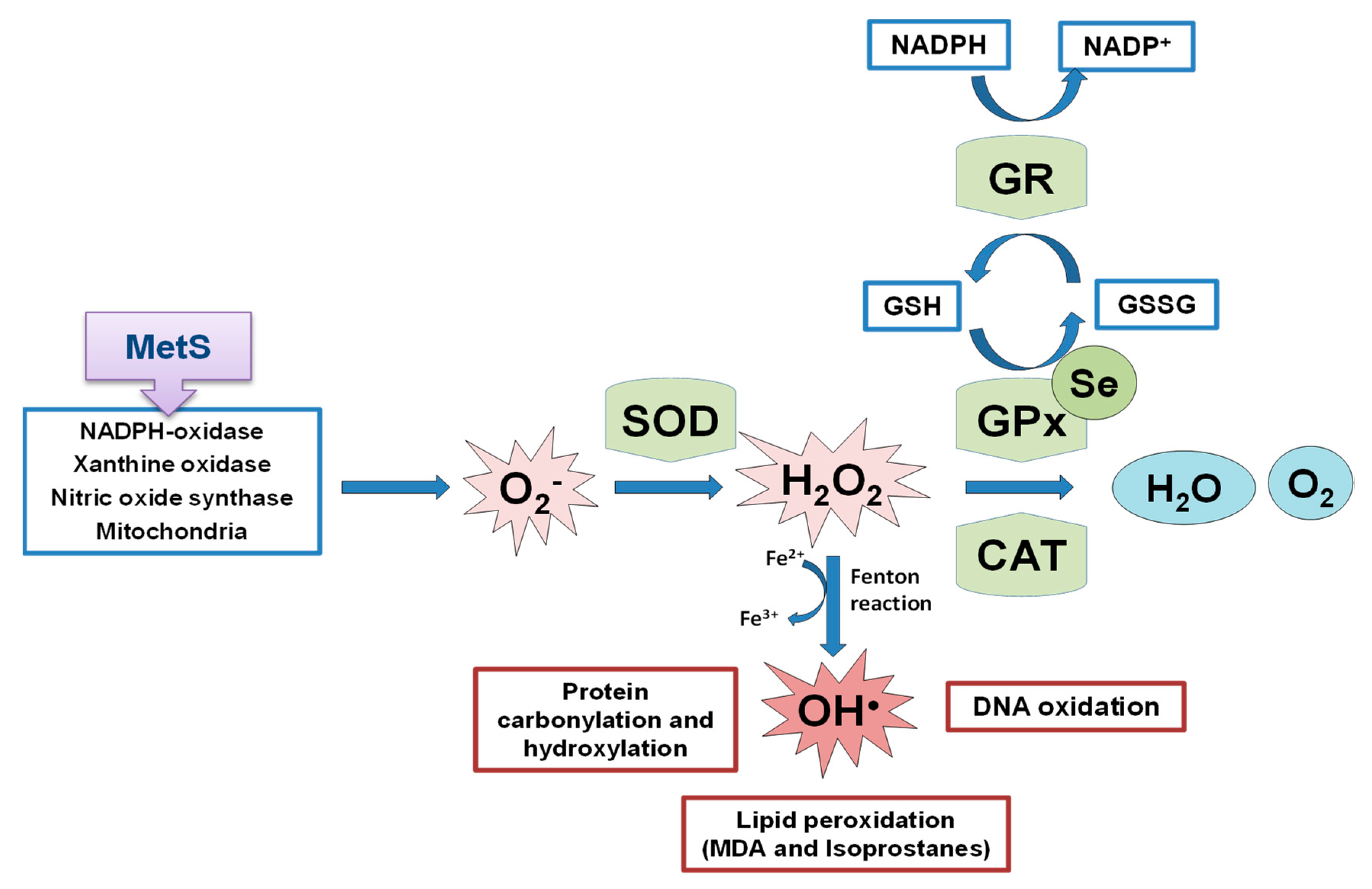 Figure 2. Schematic representation of endogenous antioxidant enzyme systems, the effect of the selenoprotein GPxs against the formed ROS. MetS implication in ROS generation. SOD: superoxide dismutase; GPx: glutathione peroxidase; CAT: catalase; GR: glutathione reductase; GSH: reduced glutathione; GSSG: oxidized glutathione; MDA: malondialdehyde.
Figure 2. Schematic representation of endogenous antioxidant enzyme systems, the effect of the selenoprotein GPxs against the formed ROS. MetS implication in ROS generation. SOD: superoxide dismutase; GPx: glutathione peroxidase; CAT: catalase; GR: glutathione reductase; GSH: reduced glutathione; GSSG: oxidized glutathione; MDA: malondialdehyde.2. Se Implications in Metabolic Programming
The theory of the origin of health and disease (the developmental origins of health and disease) proposes that the homeostatic system affected during gestational and early postnatal development impedes the ability to regulate body weight after birth, particularly with regard to high energy intake, resulting in adult obesity and metabolic diseases [95][69]. These stimuli lead to a mismatch between prenatal and/or neonatal metabolic programming, which induces an increased risk of disease in adulthood [96][70]. According to that, fetal programming occurs when the optimal environment in which the fetus grows is disrupted by insults during prenatal development, inducing changes in the metabolic state and the susceptibility of adults to develop several chronic diseases such as metabolic dysfunctions and CVD [97,98][71][72]. Therefore, fetal programming is a well-known term that relates intrauterine nutrition to the development of diseases in adult life. Early postnatal programming term includes the postnatal period where the newborn presents a very active and rapid growth, which is also influenced by environmental factors, such as the breastfeeding period. The term suggests that nutritional changes during this neonatal period lead to a greater risk of disease later in life.
OS is one of the fundamental insults related to adverse fetal programming outcomes, leading to intrauterine growth retardation (IUGR) and abnormal tissue development, affecting gestational parameters, as well as endocrine metabolic balance and pregnancy disorders [99][73]. The embryo is highly susceptible to oxidative damage since the environment that surrounds it is poor in oxygen and has low antioxidant capacity. In addition, in the placenta there are numerous changes at the oxidative level, such as an increase in NOX and antioxidants, affecting the exchange of oxygen between mother and fetus and thus producing abnormalities due to hypoxia [100][74]. During the early postnatal programming (breastfeeding period), OS also plays an important role in mothers and neonates, since it compromises an appropriate lactation process which leads to growth retardation [36,101,102,103][36][75][76][77]. In this contHerext, studi, researches in humans and animals have revealed that the antioxidant Se is essential for maternal health and offspring development during reproductive periods.
3. Se Homeostasis in MetS Programming
MetS dams have normal Se serum values during the whole experimental process, despite the fact that during lactation they ingested a lower amount of food and therefore of Se [138][78]. They excreted less Se by urine, probably trying to maintain normal serum and milk Se levels. This effort was insufficient, and Se retention decreased in MetS dams, as it was found when the apparent Se balance was measured. Se deposits in the liver and kidneys were elevated in MetS dams, whereas in the heart and muscle there was a significant depletion. This shows that Se distribution during MetS in lactating dams is controversial. It is well established that HFruD produces OS through ROS formation, especially in the liver and kidneys [143,144][79][80]. Thus, the Se increase in these tissues could enhance GPx enzyme activity, protecting them from oxidation. However, this is a double-edged sword in MetS, since it has been demonstrated that overproduction of the selenoproteins GPx and SelP in the liver produces IR, Gluc intolerance, and dyslipidemia [46]. The depletion of Se found in the heart shows, for the first time, that heart Se deposits could play a primordial role in the myocardial dysfunction found in dams with MetS; since Se deficiency contributes to fibrosis and diastolic dysfunction development [145][81]. Skeletal muscle is one of the greatest Se-storing organs [146][82]; in MetS dams, its Se stores are being destroyed in order to increase Se levels in other tissues. This Se depletion could increase ROS production and contribute to disrupting the insulin signaling pathway since skeletal muscle is the largest insulin-sensitive tissue in the body [147][83]. Regarding Se homeostasis in offspring at the end of weaning, the pups from MetS dams received less Se via milk, but their serum Se levels were unaltered [140][84]. MetS-exposed pups excreted less Se via feces and urine, trying to retain this element. However, this effort did not work, because we found, for the first time, that tissue Se deposits were altered in MetS-exposed pups. Like happened in their dams, both genders of pups showed lower levels of Se in the heart and muscle, and higher levels in the kidney, pancreas, and thyroid. However, only female pups presented a significant repletion of Se in the liver, having the same pattern of Se distribution as their mothers. Accordingly, this shows that this behavior could be typical in females. In conclusion, it has been shown that maternal MetS causes changes in Se tissue deposits of suckling pups. These changes will contribute to different tissues’ selenoprotein expression and to alterations in tissues’ oxidative balance and function, contributing to CVD, endocrine alterations, and IUGR.References
- Rayman, M.P. The importance of selenium to human health. Lancet 2000, 356, 233–241.
- Hariharan, S.; Dharmaraj, S. Selenium and selenoproteins: It’s role in regulation of inflammation. Inflammopharmacology 2020, 28, 667–695.
- Maiyo, F.; Singh, M. Selenium nanoparticles: Potential in cancer gene and drug delivery. Nanomedicine 2017, 12, 1075–1089.
- Rayman, M.P. Selenium and human health. Lancet 2012, 379, 1256–1268.
- Avery, J.C.; Hoffmann, P.R. Selenium, Selenoproteins, and Immunity. Nutrients 2018, 10, 1203.
- Fontenelle, L.C.; Feitosa, M.M.; Freitas, T.E.C.; Severo, J.S.; Morais, J.B.S.; Henriques, G.S.; Oliveira, F.E.; Moita Neto, J.M.; Marreiro, D.; Do, N. Selenium status and its relationship with thyroid hormones in obese women. Clin. Nutr. ESPEN 2021, 41, 398–404.
- Chen, L.-L.; Huang, J.-Q.; Wu, Y.-Y.; Chen, L.-B.; Li, S.-P.; Zhang, X.; Wu, S.; Ren, F.-Z.; Lei, X.-G. Loss of Selenov predisposes mice to extra fat accumulation and attenuated energy expenditure. Redox Biol. 2021, 45, 102048.
- Sneddon, A.A. Selenium and vascular health. Pure Appl. Chem. 2011, 84, 239–248.
- Nazıroğlu, M.; Öz, A.; Yıldızhan, K. Selenium and Neurological Diseases: Focus on Peripheral Pain and TRP Channels. Curr. Neuropharmacol. 2020, 18, 501–517.
- Rayman, M.P.; Winther, K.H.; Pastor-Barriuso, R.; Cold, F.; Thvilum, M.; Stranges, S.; Guallar, E.; Cold, S. Effect of long-term selenium supplementation on mortality: Results from a multiple-dose, randomised controlled trial. Free Radic. Biol. Med. 2018, 127, 46–54.
- Rayman, M.P. Selenium intake, status, and health: A complex relationship. Hormones 2020, 19, 9–14.
- Zachariah, M.; Maamoun, H.; Milano, L.; Rayman, M.P.; Meira, L.B.; Agouni, A. Endoplasmic reticulum stress and oxidative stress drive endothelial dysfunction induced by high selenium. J. Cell. Physiol. 2021, 236, 4348–4359.
- Carreras, O.; Ojeda, M.L.; Nogales, F. Selenium Dietary Supplementation and Oxidative Balance in Alcoholism. In Molecular Aspects of Alcohol and Nutrition: A Volume in the Molecular Nutrition Series; Elsevier Inc.: Amsterdam, The Netherlands, 2016; pp. 133–142. ISBN 9780128010037.
- Galmés, S.; Serra, F.; Palou, A. Current state of evidence: Influence of nutritional and nutrigenetic factors on immunity in the COVID-19 pandemic framework. Nutrients 2020, 12, 2738.
- Ha, H.Y.; Alfulaij, N.; Berry, M.J.; Seale, L.A. From Selenium Absorption to Selenoprotein Degradation. Biol. Trace Elem. Res. 2019, 192, 26.
- Schrauzer, G.N. Selenomethionine: A Review of Its Nutritional Significance, Metabolism and Toxicity. J. Nutr. 2000, 130, 1653–1656.
- Shetty, S.; Marsicano, J.R.; Copeland, P.R. Uptake and utilization of selenium from Selenoprotein P. Biol. Trace Elem. Res. 2018, 181, 54.
- Delgado, M.J.; Nogales, F.; Ojeda, M.L.; Murillo, M.L.; Carreras, O. Effect of dietary selenite on development and intestinal absorption in offspring rats. Life Sci. 2011, 88, 150–155.
- Wolffram, S.; Grenacher, B.; Scharrer, E. Transport of Selenate and Sulphate across the Intestinal Brush-Border Membrane of Pig Jejunum by Two Common Mechanisms. Q. J. Exp. Physiol. 1988, 73, 103–111.
- Thiry, C.; Ruttens, A.; Pussemier, L.; Schneider, Y.J. An in vitro investigation of species-dependent intestinal transport of selenium and the impact of this process on selenium bioavailability. Br. J. Nutr. 2013, 109, 2126–2134.
- Kato, T.; Read, R.; Rozga, J.; Burk, R.F.; Kato, R.; Read, J.; Rozga, R.; Burk, F. Evidence for intestinal release of absorbed selenium in a form with high hepatic extraction. Am. J. Physiol. Liver Physiol. 1992, 262, G854–G858.
- Lyons, M.P.; Papazyan, T.T.; Surai, P.F. Selenium in food chain and animal nutrition: Lessons from nature-review. Asian-Australas. J. Anim. Sci. 2007, 20, 1135–1155.
- Turrubiates-Hernández, F.J.; Márquez-Sandoval, Y.F.; González-Estevez, G.; Reyes-Castillo, Z.; Muñoz-Valle, J.F. The relevance of selenium status in rheumatoid arthritis. Nutrients 2020, 12, 3007.
- Kang, D.; Lee, J.; Wu, C.; Guo, X.; Lee, B.J.; Chun, J.S.; Kim, J.H. The role of selenium metabolism and selenoproteins in cartilage homeostasis and arthropathies. Exp. Mol. Med. 2020, 52, 1198–1208.
- Saito, Y. Selenoprotein P as a significant regulator of pancreatic β cell function. J. Infect. Dis. 2019, 220, 119–124.
- Burk, R.F.; Hill, K.E. Regulation of Selenium Metabolism and Transport. Annu. Rev. Nutr. 2015, 35, 109–134.
- Ogra, Y.; Anan, Y. Selenometabolomics: Identification of selenometabolites and specification of their biological significance by complementary use of elemental and molecular mass spectrometry. J. Anal. At. Spectrom. 2009, 24, 1477–1488.
- Solovyev, N.; Berthele, A.; Michalke, B. Selenium speciation in paired serum and cerebrospinal fluid samples. Anal. Bioanal. Chem. 2013, 405, 1875–1884.
- Solovyev, N.; Drobyshev, E.; Blume, B.; Michalke, B. Selenium at the Neural Barriers: A Review. Front. Neurosci. 2021, 15, 88.
- Burk, R.F.; Hill, K.E.; Olson, G.E.; Weeber, E.J.; Motley, A.K.; Winfrey, V.P.; Austin, L.M. Deletion of Apolipoprotein E Receptor-2 in Mice Lowers Brain Selenium and Causes Severe Neurological Dysfunction and Death When a Low-Selenium Diet is Fed. J. Neurosci. 2007, 27, 6207.
- Nogales, F.; Ojeda, M.L.; Fenutría, M.; Murillo, M.L.; Carreras, O. Role of selenium and glutathione peroxidase on development, growth, and oxidative balance in rat offspring. Reproduction 2013, 146, 659–667.
- Ojeda, M.L.; Carreras, O.; Díaz-Castro, J.; Murillo, M.L.; Nogales, F. High- and low- selenium diets affect endocrine energy balance during early programming. Toxicol. Appl. Pharmacol. 2019, 382, 114744.
- Sunde, R.A.; Thompson, K.M. Dietary selenium requirements based on tissue selenium concentration and glutathione peroxidase activities in old female rats. J. Trace Elem. Med. Biol. 2009, 23, 132–137.
- Thisse, C.; Degrave, A.; Kryukov, G.V.G.V.; Gladyshev, V.N.; Obrecht-Pflumio, S.; Krol, A.; Thisse, B.; Lescure, A. Spatial and temporal expression patterns of selenoprotein genes during embryogenesis in zebrafish. Gene Expr. Patterns 2003, 3, 525–532.
- Roman, M.; Jitaru, P.; Barbante, C. Selenium biochemistry and its role for human health. Metallomics 2013, 6, 25–54.
- Ojeda, M.L.; Nogales, F.; Romero-Herrera, I.; Carreras, O. Fetal Programming is Deeply Related to Maternal Selenium Status and Oxidative Balance; Experimental Offspring Health Repercussions. Nutrients 2021, 13, 2085.
- Kasaikina, M.V.; Hatfield, D.L.; Gladyshev, V.N. Understanding selenoprotein function and regulation through the use of rodent models. Biochim. Biophys. Acta-Mol. Cell Res. 2012, 1823, 1633–1642.
- Ojeda, L.; Nogales, F.; Murillo, L.; Carreras, O. The role of folic acid and selenium against oxidative damage from ethanol in early life programming: A review. Biochem. Cell Biol. 2018, 96, 178–188.
- Qazi, I.H.; Angel, C.; Yang, H.; Pan, B.; Zoidis, E.; Zeng, C.J.; Han, H.; Zhou, G. Bin Selenium, selenoproteins, and female reproduction: A review. Molecules 2018, 23, 3053.
- Khurana, A.; Tekula, S.; Saifi, M.A.; Venkatesh, P.; Godugu, C. Therapeutic applications of selenium nanoparticles. Biomed. Pharmacother. 2019, 111, 802–812.
- Lu, J.; Holmgren, A. Selenoproteins. J. Biol. Chem. 2009, 284, 723–727.
- Zoidis, E.; Seremelis, I.; Kontopoulos, N.; Danezis, G.; Zoidis, E.; Seremelis, I.; Kontopoulos, N.; Danezis, G.P. Selenium-Dependent Antioxidant Enzymes: Actions and Properties of Selenoproteins. Antioxidants 2018, 7, 66.
- McKenzie, R.C.; Arthur, J.R.; Beckett, G.J. Selenium and the Regulation of Cell Signaling, Growth, and Survival: Molecular and Mechanistic Aspects. Antioxid. Redox Signal. 2004, 4, 339–351.
- Stapleton, S.R.; Garlock, G.L.; Foellmi-Adams, L.; Kletzien, R.F. Selenium: Potent stimulator of tyrosyl phosphorylation and activator of MAP kinase. Biochim. Biophys. Acta-Mol. Cell Res. 1997, 1355, 259–269.
- Steinbrenner, H.; Speckmann, B.; Pinto, A.; Sies, H. High selenium intake and increased diabetes risk: Experimental evidence for interplay between selenium and carbohydrate metabolism. J. Clin. Biochem. Nutr. 2011, 48, 40–45.
- Steinbrenner, H. Interference of selenium and selenoproteins with the insulin-regulated carbohydrate and lipid metabolism. Free Radic. Biol. Med. 2013, 65, 1538–1547.
- Pinto, A.; Speckmann, B.; Heisler, M.; Sies, H.; Steinbrenner, H. Delaying of insulin signal transduction in skeletal muscle cells by selenium compounds. J. Inorg. Biochem. 2011, 105, 812–820.
- Wang, X.; Zhang, W.; Chen, H.; Liao, N.; Wang, Z.; Zhang, X.; Hai, C. High selenium impairs hepatic insulin sensitivity through opposite regulation of ROS. Toxicol. Lett. 2014, 224, 16–23.
- Schomburg, L. Selenium Deficiency Due to Diet, Pregnancy, Severe Illness, or COVID-19—A Preventable Trigger for Autoimmune Disease. Int. J. Mol. Sci. 2021, 22, 8532.
- Steinbrenner, H.; Duntas, L.H.; Rayman, M.P. The role of selenium in type-2 diabetes mellitus and its metabolic comorbidities. Redox Biol. 2022, 50, 102236.
- Misu, H.; Takamura, T.; Takayama, H.; Hayashi, H.; Matsuzawa-Nagata, N.; Kurita, S.; Ishikura, K.; Ando, H.; Takeshita, Y.; Ota, T.; et al. A liver-derived secretory protein, selenoprotein P, causes insulin resistance. Cell Metab. 2010, 12, 483–495.
- Brigelius-Flohé, R.; Flohé, L. Selenium and redox signaling. Arch. Biochem. Biophys. 2017, 617, 48–59.
- Brigelius-Flohé, R.; Maiorino, M. Glutathione peroxidases. Biochim. Biophys. Acta-Gen. Subj. 2013, 1830, 3289–3303.
- Ojeda, M.L.; Carreras, O.; Muñoz del Valle, P.; Murillo, M.L.; Nogales, F. Fructose exposure during gestation and lactation altered hepatic selenoprotein expression, oxidative balance and metabolic profile in female rat pups. J. Funct. Foods 2018, 43.
- Misu, H. Pathophysiological significance of hepatokine overproduction in type 2 diabetes. Diabetol. Int. 2018, 9, 224–233.
- Stapleton, S.R. Selenium: An insulin-mimetic. Cell. Mol. Life Sci. CMLS 2000, 57, 1874–1879.
- Rayman, M.P.; Stranges, S. Epidemiology of selenium and type 2 diabetes: Can we make sense of it? Free Radic. Biol. Med. 2013, 65, 1557–1564.
- Seale, L.A.; Hashimoto, A.C.; Kurokawa, S.; Gilman, C.L.; Seyedali, A.; Bellinger, F.P.; Raman, A.V.; Berry, M.J. Disruption of the selenocysteine lyase-mediated selenium recycling pathway leads to metabolic syndrome in mice. Mol. Cell. Biol. 2012, 32, 4141–4154.
- Kremer, P.M.; Torres, D.J.; Hashimoto, A.C.; Berry, M.J. Sex-Specific Metabolic Impairments in a Mouse Model of Disrupted Selenium Utilization. Front. Nutr. 2021, 8.
- Watanabe, L.M.; Hashimoto, A.C.; Torres, D.J.; Berry, M.J.; Seale, L.A. Effects of selenium supplementation on diet-induced obesity in mice with a disruption of the selenocysteine lyase gene. J. Trace Elem. Med. Biol. 2020, 62.
- Seale, L.A.; Gilman, C.L.; Hashimoto, A.C.; Ogawa-Wong, A.N.; Berry, M.J. Diet-induced obesity in the selenocysteine lyase knockout mouse. Antioxid. Redox Signal. 2015, 23, 761–774.
- Daenen, K.; Andries, A.; Mekahli, D.; Van Schepdael, A.; Jouret, F.; Bammens, B. Oxidative stress in chronic kidney disease. Pediatr. Nephrol. 2019, 34, 975–991.
- Shao, D.; Oka, S.I.; Brady, C.D.; Haendeler, J.; Eaton, P.; Sadoshima, J. Redox modification of cell signaling in the cardiovascular system. J. Mol. Cell. Cardiol. 2012, 52, 550–558.
- Vona, R.; Gambardella, L.; Cittadini, C.; Straface, E.; Pietraforte, D. Biomarkers of Oxidative Stress in Metabolic Syndrome and Associated Diseases. Oxid. Med. Cell. Longev. 2019, 2019.
- Torres-Cuevas, I.; Parra-Llorca, A.; Sánchez-Illana, A.; Nuñez-Ramiro, A.; Kuligowski, J.; Cháfer-Pericás, C.; Cernada, M.; Escobar, J.; Vento, M. Oxygen and oxidative stress in the perinatal period. Redox Biol. 2017, 12, 674.
- Rani, V.; Deep, G.; Singh, R.K.; Palle, K.; Yadav, U.C.S. Oxidative stress and metabolic disorders: Pathogenesis and therapeutic strategies. Life Sci. 2016, 148, 183–193.
- Senoner, T.; Dichtl, W. Oxidative stress in cardiovascular diseases: Still a therapeutic target? Nutrients 2019, 11, 2090.
- Younus, H. Therapeutic potentials of superoxide dismutase. Int. J. Health Sci. 2018, 12, 88.
- Barker, D. The developmental origins of chronic adult disease. Acta Paediatr. Suppl. 2004, 93, 26–33.
- Moreno-Mendez, E.; Quintero-Fabian, S.; Fernandez-Mejia, C.; Lazo-De-La-Vega-Monroy, M.L. Early-life programming of adipose tissue. Nutr. Res. Rev. 2020, 33, 244–259.
- Thompson, L.P.; Al-Hasan, Y. Impact of oxidative stress in fetal programming. J. Pregnancy 2012, 2012.
- Perrone, S.; Santacroce, A.; Picardi, A.; Buonocore, G. Fetal programming and early identification of newborns at high risk of free radical-mediated diseases. World J. Clin. Pediatr. 2016, 5, 172.
- Rana, S.; Lemoine, E.; Granger, J.; Karumanchi, S.A. Preeclampsia: Pathophysiology, Challenges, and Perspectives. Circ. Res. 2019, 124, 1094–1112.
- Schoots, M.H.; Gordijn, S.J.; Scherjon, S.A.; van Goor, H.; Hillebrands, J.L. Oxidative stress in placental pathology. Placenta 2018, 69, 153–161.
- Zheng, G.X.; Lin, J.T.; Zheng, W.H.; Cao, J.; Zhao, Z.J. Energy intake, oxidative stress and antioxidant in mice during lactation. Zool. Res. 2015, 36, 95–102.
- Sordillo, L.M. Selenium-dependent regulation of oxidative stress and immunity in periparturient dairy cattle. Vet. Med. Int. 2013, 2013.
- Pinedo, P.J.; De Vries, A. Effect of days to conception in the previous lactation on the risk of death and live culling around calving. J. Dairy Sci. 2010, 93, 968–977.
- Nogales, F.; Ojeda, M.L.; del Valle, P.M.; Serrano, A.; Murillo, M.L.; Carreras Sánchez, O. Metabolic syndrome and selenium during gestation and lactation. Eur. J. Nutr. 2017, 56, 819–830.
- Sreeja, S.; Geetha, R.; Priyadarshini, E.; Bhavani, K.; Anuradha, C.V. Substitution of Soy Protein for Casein Prevents Oxidative Modification and Inflammatory Response Induced in Rats Fed High Fructose Diet. ISRN Inflamm. 2014, 2014, 1–8.
- Reddi, A.S.; Bollineni, J.S. Selenium-deficient diet induces renal oxidative stress and injury via TGF-β1 in normal and diabetic rats. Kidney Int. 2001, 59, 1342–1353.
- Metes-Kosik, N.; Luptak, I.; DiBello, P.M.; Handy, D.E.; Tang, S.-S.; Zhi, H.; Qin, F.; Jacobsen, D.W.; Loscalzo, J.; Joseph, J. Both Selenium Deficiency and Modest Selenium Supplementation Lead to Myocardial Fibrosis in Mice via Effects on Redox-Methylation Balance. Mol. Nutr. Food Res. 2012, 56, 1812.
- Daniels, L.A. Selenium metabolism and bioavailability. Biol. Trace Elem. Res. 1996, 54, 185–199.
- Stump, C.S.; Henriksen, E.J.; Wei, Y.; Sowers, J.R. The metabolic syndrome: Role of skeletal muscle metabolism. Ann. Med. 2009, 38, 389–402.
- Ojeda, M.L.; Nogales, F.; Muñoz Del Valle, P.; Díaz-Castro, J.; Murillo, M.L.; Carreras, O. Metabolic syndrome and selenium in fetal programming: Gender differences. Food Funct. 2016, 7, 3031–3038.
More