Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Camila Xu and Version 1 by Martin Barr.
Natural killer (NK) cells were first described in the 1970's and belong to a subgroup of the innate lymphoid cell family. They arise from common lymphoid progenitors, but unlike T cells and B cells, they lack genetically rearranged receptors, are independent of antigen specificity and rely on a balance of signals transduced via activating and inhibitory receptors to induce activation.
- NSCLC
- NK cells
- NK cell therapies
- immunotherapy
- lung cancer
[1][2][3][4][5][6][7][8][9][10][11][12][13][14][15][16][17][18][19][20][21][22][23][24][25][26][27][28][29][30][31][32][33][34][35][36][37][38][39][40][41][42][43][44][45][46][47][48][49][50][51][52][53][54][55][56][57][58][59][60][61][62][63][64][65][66][67][68][69][70][71][72][73][74][75][76][77][78][79]1. Introduction
Cancer is a leading cause of death worldwide, with over 10 million deaths reported in 2020 alone. Lung cancer was only recently surpassed as the most common cancer type, yet it remains the primary cause of cancer-related mortality, with almost 1.8 million individuals succumbing to the disease [1]. Lung cancer is classified as either small cell (SCLC) or non-small cell lung cancer (NSCLC). NSCLC accounts for approximately 85% of all lung cancer diagnoses and can be further subdivided into several subtypes: adenocarcinoma (ADC), squamous cell carcinoma (SqCC), large cell carcinoma (LCC), and mixed histology [2]. ADC arises from alveolar cells and represents approximately 40% of NSCLC cases. Conversely, SqCC arises from the bronchial epithelium and accounts for 25–30% of diagnoses [3]. The average 5-year survival rate for NSCLC varies and is dependent on cancer stage; rates range, from 63% for Stage I NSCLC, to a dismal 7% for Stage IV NSCLC [4]. While early stage lung cancer, for most patients, is resectable with curative intent, approximately 70% of patients present with advanced stage disease [5].
2. Clinical Challenges Associated with Non-Small Cell Lung Cancer
Cigarette smoking, current or former, is the most common risk factor associated with the development of lung cancer and is responsible for approximately 80% of cases [6]. SqCC is more commonly associated with smoking and chronic inflammation in contrast to ADC, which is becoming increasingly diagnosed in never-smokers and tends to occur more often in younger females [7]. Smokers tend to have higher mutation frequencies in Kirsten rat sarcoma (KRAS) and tumor protein P53 (TP53), whereas higher levels of actionable mutations, such as epidermal growth factor receptor (EGFR), ros-oncogene 1 (ROS1), and anaplastic lymphoma kinase (ALK), are associated with never-smokers [8]. Other risk factors include the use of other tobacco products, exposure to second-hand smoke, occupational carcinogens, radiation, radon, and air pollution [6].
In terms of treatment modalities, surgical resection with curative intent is the most effective treatment for stage I-IIIA NSCLC [3]. Patients can also be offered a combination of radiation therapy, chemotherapy, and/or surgery, depending on variables such as tumor size, location, lymph node involvement, underlying co-morbidities, and tolerance to treatment [9]. Immunotherapies can also be administered, as either first-line treatment or as an adjuvant therapy [9]. In advanced disease, where targeted or immunotherapies are not possible, platinum-based chemotherapy such as cisplatin is often used [10]. However, cisplatin resistance has become a significant clinical challenge in these patient cohorts.
NSCLC is a molecularly heterogeneous disease, comprising various genetic abnormalities that further subdivide the disease into specific molecular subtypes. EGFR mutations are one of the most common alterations observed in NSCLC and occur most frequently as either exon 19 deletions or exon 21 L858R mutations [11]. FDA-approved targeted treatments for EGFR mutations include the tyrosine kinase inhibitors erlotinib, gefitinib, afatinib, osimertinib, and dacomitinib. In advanced stage disease, most of these therapies are administered as monotherapies. However, erlotinib can be used in combination with angiogenesis inhibitors such as bevacizumab and ramucircumab in advanced stage NSCLC, while osimertinib can be used as an adjuvant treatment post-surgery for early-stage NSCLC [12,13][12][13]. These therapies are not as effective in patients with EGFR exon 20 insertion mutations; however, the FDA has recently approved therapies for targeting this mutation, including mobocertinib and amivantamab, which are typically administered after failure of prior platinum based-chemotherapy [12,13][12][13].
Mutations in KRAS are also frequently observed in NSCLC. Approximately 13% of patients present with a specific mutation, KRAS G12C. This mutation is often resistant to other targeted therapies such as EGFR inhibitors [13]. Sotorasib received FDA-approval in June 2021 as a first-line treatment for individuals with this genetic mutation after treatment of at least one prior systemic therapy [12]. These aforementioned genetic mutations are most frequently observed in ADC alongside mutations in tumor-suppressor genes TP53, Kelch-like ECH-associated protein 1 (KEAP1), serine/threonine kinase 11 (STK11), and neurofibromin 1 (NF1) [8]. Less common genetic mutations include mutations in ALK, ROS1, B-Raf proto-oncogene serine/threonine kinase (BRAF), rearranged during transfection proto-oncogene (RET), and mesenchymal epithelial transition (MET). Approximately 5% of NSCLC diagnoses are EML4-ALK-positive, where this fusion protein is commonly observed in young patients and non-smokers [13]. Loratinib is the first-line treatment for this subtype, but additional therapies including crizotinib, certinib, alectinib, and brigatinib are also used [12,13][12][13]. Mutations in ROS1 are rare, but the gene rearrangement shares similarities with ALK gene rearrangements. Therapies that can be administered for ALK mutations include crizotinib, certinib, and lorlatinib, which act by inhibiting ALK enzyme activity [14]. Entrectinib can be administered for individuals with metastatic NSCLC with ALK mutations. Therapies targeting BRAF mutations include dabrafenib and trametinib, which can be used in combination for metastatic NSCLC. RET inhibitors such as selpercatinib and pralsetinib target the abnormal RET protein, while MET inhibitors such as capmatinib and tepotinib can be used to treat metastatic NSCLC characterized by MET gene mutations [13]. However, these therapies continue to present a significant clinical challenge, in that they are only effective in individuals harboring actionable mutations, after which time, drug resistance eventually develops. As observed with targeted therapies, treatment options for NSCLC have evolved from a ‘one size fits all’ approach to a more personalized strategy based on the specific tumor characteristics.
The introduction of immunotherapies has revolutionized the cancer landscape, improving response and survival rates for patients with advanced stage NSCLC [15]. Current immune checkpoint inhibitors (ICIs) have the ability to target several immune checkpoint signals, including the classical programmed cell death protein 1 (PD-1), programmed cell death ligand 1 (PD-L1), and cytotoxic T lymphocyte-associated protein (CTLA-4) checkpoints, in addition to new emerging checkpoints such as T cell immunoglobulin and mucin domain-containing protein 3 (TIM-3), lymphocyte activation gene-3 (LAG-3) and T cell immunoreceptor with Ig and ITIM domains (TIGIT) [15]. Anti-PD-1/PD-L1 therapies have demonstrated efficacy in many malignancies in NSCLC. Anti-PD-1 monoclonal antibodies (mAbs) such as nivolumab, pembrolizumab and cemiplimab, and anti-PD-L1 mAbs atezolizumab and durvalumab target PD-1/PD-L1 binding function, by essentially removing the brakes from the anti-tumor immune response [13]. These immunotherapies are offered as a first-line therapy for NSCLC or in conjunction with other immunotherapies or chemotherapy [13]. Despite their effectiveness, ICIs are not a panacea and do not fully address the treatment requirements of all NSCLC patients. It has been shown that up to 30% of patients show rapid progression, while only 10–15% of patients demonstrate long term benefits [16]. In addition, the use of ICIs leads to immunotherapy-induced resistance, presenting another significant clinical challenge in NSCLC [17]. The combination of multiple ICIs has also demonstrated improved clinical responses. In a review by Ma et al., the presence of other untargeted immune checkpoints was highlighted, possibly compensating for the blockade of specific signals [15]. This provides a rationale for combining ICIs, and many trials have investigated the utility of this combinatorial approach and have found its superiority over monotherapy [18,19][18][19].
3. Natural Killer Cells in Health and Disease
Natural killer (NK) cells were first described in the 1970s and belong to a subgroup of the innate lymphoid cell family [21][20]. They arise from common lymphoid progenitors, but unlike T cells and B cells, they lack genetically rearranged receptors, are independent of antigen specificity and rely on a balance of signals transduced via activating and inhibitory receptors to induce activation [22][21]. NK cells represent 5–15% of circulating lymphocytes and are present primarily in the bloodstream and in lymphatic vessels [23,24][22][23]. They also act as sentinels in various other sites, such as the liver, bone marrow, and the lungs [23][22]. The heterogenous NK cell population is commonly and broadly characterized based on their maturation status and expression levels of CD56 and CD16 [23,25][22][24]. Approximately 90% of all peripheral blood NK cells are classified as CD56dimCD16bright. This subset of NK cells are highly cytotoxic, produce modest levels of cytokines, and express markers such as CD16, CD57, and PEN5. By contrast, the CD56brightCD16- subset mainly reside in secondary lymphoid organs and express markers such as CD122, NKp46, and NKp80. These CD56bright NK cells produce a larger number of cytokines and chemokines compared to the CD56dim subset and are less cytotoxic [26,27,28][25][26][27]. It has been shown that following activation, the immature CD56bright NK cells increase their expression of NK receptors characteristic of the more mature and cytotoxic CD56dim subset [29,30,31][28][29][30].
NK cells form part of the first line of defense in mediating viral infections and cancer immune surveillance, particularly cancer metastases. They exercise their cytotoxic effects without the need for pre-activation via the release of perforin and granzymes [32][31]. The cytotoxic functions of NK cells are induced by a delicate interplay between activating and inhibitory NK cell receptors (Figure 1) and their cell-surface bound ligands (Table 1). Inhibitory receptors such as killer cell immunoglobulin-like receptors (KIRs) and the CD94-NKG2A complex have the ability to recognize self-molecules on the surface of normal cells and inhibit NK cell activation [26,33][25][32]. In addition, the immunoreceptor tyrosine-based inhibitory motifs (ITIMs) of certain inhibitory receptors can interact with MHC class I molecules to aid in the processes of NK cell licensing and education, ensuring full activation and self-tolerance of NK cells [26][25].
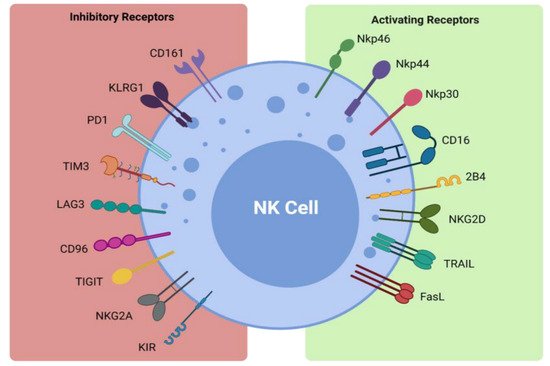
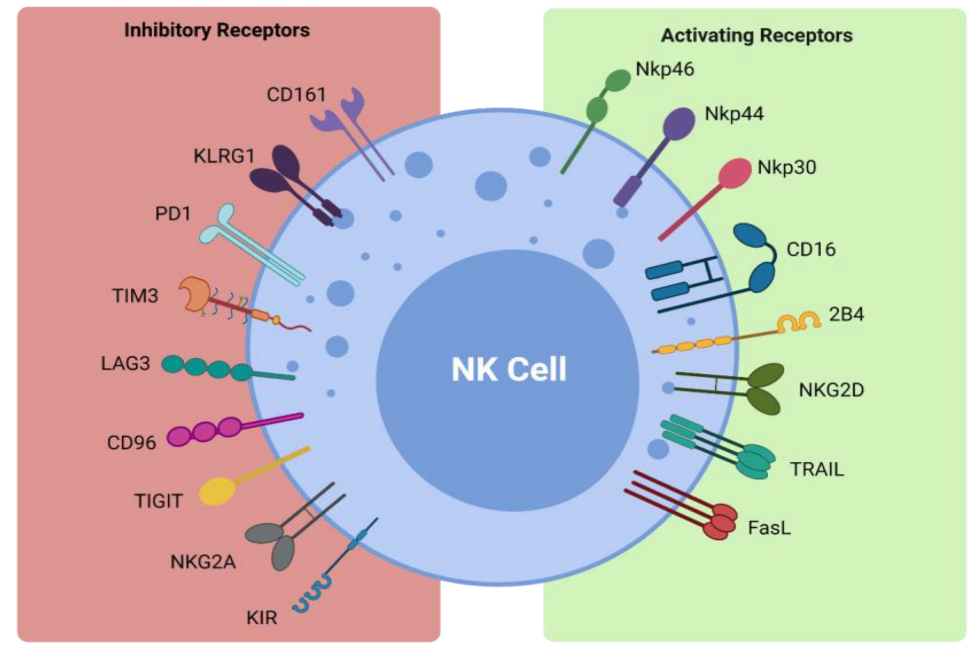 Figure 1. Activating and inhibitory NK cell receptors. Figure illustrating the panel of inhibitory and activating receptors expressed on the surface of NK cells that regulate their activation and effector function (www.biorender.com, accessed 21 December 2021).
Figure 1. Activating and inhibitory NK cell receptors. Figure illustrating the panel of inhibitory and activating receptors expressed on the surface of NK cells that regulate their activation and effector function (www.biorender.com, accessed 21 December 2021).| Receptor | Ligand |
|---|
| Inhibitory Receptors and Ligands | |
| CD161 | LLT1 |
| KLRG1 | Cadherins |
| PD-1 | PD-L1 |
| TIM3 | Galectin 9, phosphatidylserine, CEACAM1, HMGB1 |
| LAG3 | MHC class II |
| CD96 | CD155 |
| TIGIT | CD155, CD112, CD113 |
| NKG2A | HLA-E |
| KIR | HLA-C/B/A |
| Activating Receptors and Ligands | |
| NKp46 | Viral hemagglutinins |
| NKp44 | Viral hemagglutinins |
| NKp30 | PP65, BAT-3 |
| CD16 | IgG |
| 2B4 | CD48 |
| NKG2D | ULBP, MICA/B |
| TRAIL | TRAIL-R1, TRAIL-R2 |
| FasL | Fas |
Activating NK cell receptors include receptors such as NKG2D and the natural cytotoxicity receptors NKp30, NKp44, and NKp46 [26,40,41,42,43][25][39][40][41][42]. They bind to stress-induced self-ligands on infected or malignant cells and trigger NK cell cytokine production and cytotoxicity, while killer activating receptors upregulate death ligands such as TNF-α, FasL, and TRAIL, which are key components in the apoptosis of target cells [32,37,38][31][36][37]. The CD16 receptor is another activating receptor that is responsible for the triggering of antibody-dependent-cellular cytotoxicity (ADCC) against antibody- coated target cells [26][25]. Infected or transformed cells often downregulate their MHC class I expression to evade detection by T cells and their absence can activate NK cells in a process coined ‘missing-self recognition’ [26][25]. NK cells can also modulate the immune response via interaction with other immune cells, such as cross-presentation of antigens from apoptotic target cells to particular subsets of dendritic cells and priming of CD4+ helper T cells via interferon-γ (IFN-γ) production, as reviewed by Vivier et al. [26,44,45][25][43][44]. Importantly, NK cells can also promote resolution of an immune response by killing activated T cells or via suppression of autoreactive B lymphocytes in vitro [26,46,47][25][45][46]. Therefore, NK cells are not only potent killers of transformed cells but also promote tumor eradication via their recruitment and activation of other arms of the immune response.
4. NK Cells in NSCLC
The majority of the NK cell population in the lung is CD56brightCD16-, exhibiting high cytokine release but low cytotoxicity [48][47]. In fact, in vivo studies have demonstrated that human lung NK cells respond poorly to activation by target cells, when compared to peripheral blood NK cells. It has been proposed that this is due to the suppressive effects associated with alveolar macrophages and soluble factors that are present in the epithelial lining of the lower respiratory tract to maintain lung homeostasis [48,49][47][48]. These human lung NK cells share several phenotypic similarities with the decidual NK (dNK) cells present in the maternal decidua during pregnancy [50][49]. Such dNK cells are involved in promoting invasion of the invasive extravillous trophoblasts, vascular remodeling, and the establishment of fetal tolerance during pregnancy via the production of various cytokines/chemokines and pro-angiogenic factors [50][49]. These dNK cells have been shown to produce several pro-angiogenic factors such as vascular endothelial growth factor (VEGF), placental growth factor (PlGF), and NKG5 and are potent secretors of IL-8 [51][50]. The CD56brightCD16dim subset that are enriched in NSCLC tumor samples have shown similarities to dNK cells, in that they produce some of the same pro-angiogenic factors, such as VEGF, PlGF, and IL-8, promoting tumor growth and metastases [52][51]. While this NK cell phenotype in the maternal decidua permits the establishment and maintenance of pregnancy and is beneficial, this NK cell subtype in NSCLC is more likely to be harmful through the promotion of tumorigenesis and certainly warrants further investigation. Multiple studies have reported that the infiltration of NK cells into solid tumors is associated with favorable prognosis in many cancers [53,54][52][53]. In lung cancer, downregulated NK cell receptor expression has been reported on intratumoral NK cells, while defective degranulation and IFN-γ production has also been observed [55][54]. Therefore, the restoration of NK cell responses using NK cell therapies is a desirable therapeutic concept in this malignancy.
Suppression and Evasion of NK Cell Responses within the NSCLC Tumor Microenvironment
The hostile tumor microenvironment (TME) can impair NK cell functions by attenuating their cytotoxic capabilities via the production of immunosuppressive factors and via nutrient deprived and hypoxic conditions [56][55] (Figure 2). Furthermore, the TME’s role in propagating pro-tumor responses presents a significant clinical challenge when designing therapeutics.
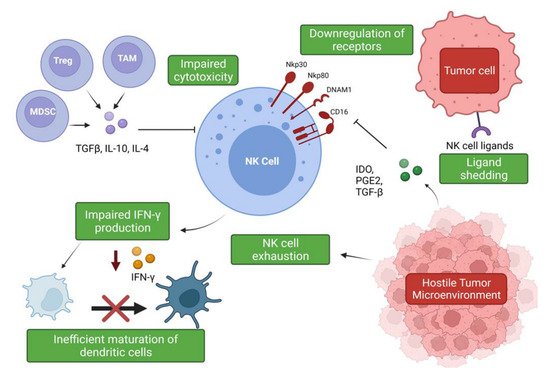
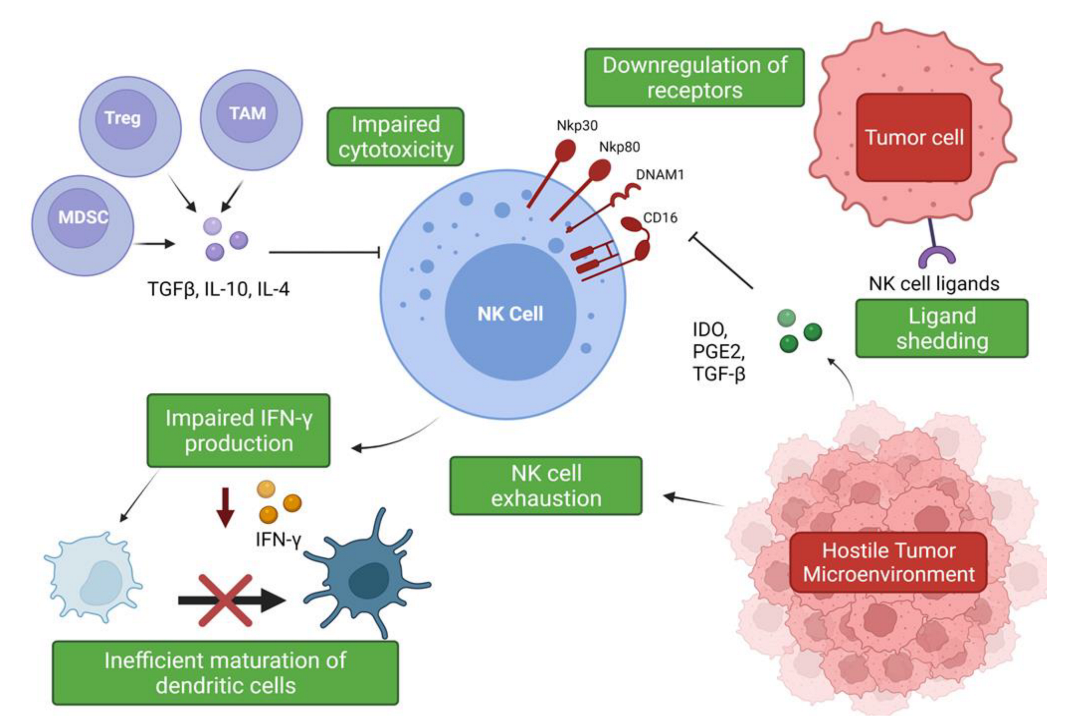 Figure 2. Suppression and evasion of NK cells in the tumor microenvironment. Figure illustrating the various factors that contribute to NK cell dysfunction in the TME and the impact on other immune cells (www.biorender.com, accessed 27 December 2021).
Figure 2. Suppression and evasion of NK cells in the tumor microenvironment. Figure illustrating the various factors that contribute to NK cell dysfunction in the TME and the impact on other immune cells (www.biorender.com, accessed 27 December 2021).The hypoxic conditions of the TME are responsible for the promotion of angiogenesis via induction of hypoxia-inducible factor-1 (HIF-1), leading to the production of pro-angiogenic factors such as VEGF [57,58][56][57]. Experimental evidence has shown that a hypoxic environment combined with TGF-β1 and Aza, a demethylating agent, enriched peripheral NK cell cultures in CD56brightCD16dim cells and induced secretion of VEGF, leading to increased angiogenesis [59][58]. Phenotypic conversion of this subset to pro-angiogenic, decidual-like NK cells has been observed in NSCLC tumor samples, with an increased production of VEGF, PlGF, and IL-8 observed in patients with both ADC and SqCC [52][51]. Exposure to TGF-β1 upregulates VEGF and P1GF production in the NK cells of healthy controls, suggesting that the pro-angiogenic phenotype associated with NSCLC is, in part, mediated by TGF-β1 [52][51]. Another member of the innate lymphoid group, type 2 innate lymphoid cells (ILC2s), has also been implicated in promoting angiogenesis and cancer progression. ILC2s have emerged as a major driver of type 2 inflammation, producing typical type 2 cytokines such as IL-4, IL-5, and IL-13 and contributing to the pathogenesis of inflammatory conditions such as asthma [60][59]. In the context of NSCLC, ILC2s have been shown to be enriched in NSCLC tumor samples and to upregulate PD-1 expression, displaying an increased expression of type 2 cytokines such as IL-4 and IL-13, suggesting these immune cells also contribute to the immunosuppressive environment [61][60]. IL-33 production driven by these cytokines is thought to be responsible for the pro-tumor activity of these immune cells, leading to increased angiogenesis and tumor metastases [62,63][61][62].
Soluble factors expressed by lung cancer cells have been associated with a decreased expression of granzyme B, perforin, and IFN-γ by infiltrating immune cells, including NK cells [64][63]. Tumor-associated macrophages (TAMs) and myeloid-derived suppressor cells (MDSCs) produce immunosuppressive factors such as TGF-β, IL-10, and PGE2, which impede NK cell cytotoxicity [27][26]. The secretion of IL-10 by TAMs suppresses NK cell cytotoxicity, and an increase in IL-10 production alongside reduced IFN-γ production has been described in patients with NSCLC [27,65][26][64]. IL-10 overproduction has been associated with enhanced angiogenesis, and, moreover, an increase in serum IL-10 levels is an indicator of poor prognosis [65,66,67][64][65][66]. However, IL-10 can significantly increase glycolysis and enhance the effector functions of NK cells via mTORC1 signaling [68][67]. These findings suggest a potential use of this classical immunomodulatory cytokine in the promotion of NK cell effector functions and warrants further investigation. TGF-β regulates NK cell function by inhibiting the expression of NKp30 and NKG2D receptors, which are vital in the recognition and destruction of tumor cells [27,69][26][68]. This inhibition has been suggested to negatively impact NK-cell mediated killing of immature dendritic cells (DC), which is an important mechanism for eliminating potentially tolerogenic DCs, in controlling the adaptive immune response [69,70][68][69]. The bi-directional cross-talk between NK cells and DCs is important for their activation and maturation, respectively [71][70]. IFN-γ production has been shown to be vital for the activation of endogenous DCs [71,72][70][71], but it has been shown that intra-tumoral NK cells are impaired in their ability to secrete IFN-γ; therefore, negatively impacting DC maturation [27,73][26][72]. It has also been shown that TGF-β drives NK cell dysfunction in the lung tumor microenvironment via FBP1 upregulation and subsequent inhibition of glycolysis in lung NK cells [74][73].
The rate-limiting enzyme involved in tryptophan catabolism, indoleamine 2,3-dioxygenase (IDO), is released by tumor cells and can inhibit the upregulation of NKp46 and NKG2D via blockade of IL-2, again limiting the NK cell’s ability to recognize and kill tumor cells [75][74]. Cigarette smoking has also been implicated in NK cell dysfunction and has been shown to reduce their cytotoxic abilities. Smokers have been shown to exhibit a significant reduction in the secretion of IFN-γ and TNF-α when compared to non-smokers. Interestingly, it was found that this inhibitory effect on TNF-α production was reversible, suggesting that smoking cessation may somewhat improve NK cell responses to cancer [76][75]. IL-15 plays an important role in the proliferation, survival, and function of NK cells, and its production was also found to be suppressed due to the effects of cigarette smoking; thereby, negatively impacting critical mechanisms of NK cell development and function [77][76]. These findings stress the need to promote smoking cessation, as it not only drives cancer development but is also a promotor of progression via impairment of the immune response. As NK cell dysfunction and impairment promotes tumor immune evasion in NSCLC, emerging therapies have focused on restoring normal function of NK cells by means of cytokine supplementation, blockade of inhibitory receptors, and neutralization of immunosuppressive cytokines such as TGF-β [73][72].
NK cells have also been shown to become exhausted as a result of the hostile TME, leading to impaired effector function and an altered phenotype [78][77]. NK cell exhaustion has been shown to be responsible for the significant decrease in NK cell cytotoxicity in lung cancer patients [79][78]. Moreover, the NKG2A receptor has been shown to be upregulated, whereas the CD226 receptor has been found to be downregulated in these patients [79][78]. The expression levels of these receptors may prove useful as markers of NK cell exhaustion and as potential therapeutic targets in this cohort.
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN.Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA A Cancer J. Clin. 2021, 71, 209–249.
- Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA A Cancer J. Clin. 2021, 71, 209–249.Griffin, R.; Ramirez, R.A. Molecular Targets in Non-Small Cell Lung Cancer. Ochsner J. 2017, 17, 388–392.
- Griffin, R.; Ramirez, R.A. Molecular Targets in Non-Small Cell Lung Cancer. Ochsner J. 2017, 17, 388–392.Duma, N.; Santana-Davila, R.; Molina, J.R. Non–Small Cell Lung Cancer: Epidemiology, Screening, Diagnosis, and Treatment. Mayo Clin. Proc. 2019, 94, 1623–1640.
- Duma, N.; Santana-Davila, R.; Molina, J.R. Non–Small Cell Lung Cancer: Epidemiology, Screening, Diagnosis, and Treatment. Mayo Clin. Proc. 2019, 94, 1623–1640.Howlader, N.; Noone, A.M.; Krapcho, M.; Miller, D.; Brest, A.; Yu, M.; Ruhl, J.; Tatalovich, Z.; Mariotto, A.; Lewis, D.R.; et al. SEER Cancer Statistics Review, 1975-2018, National Cancer Institute. Available online: https://seer.cancer.gov/csr/1975_2018/ (accessed on 19 December 2021).
- Howlader, N.; Noone, A.M.; Krapcho, M.; Miller, D.; Brest, A.; Yu, M.; Ruhl, J.; Tatalovich, Z.; Mariotto, A.; Lewis, D.R.; et al. SEER Cancer Statistics Review, 1975-2018, National Cancer Institute. Available online: https://seer.cancer.gov/csr/1975_2018/(accessed on 19 December 2021).Cagle, P.T.; Allen, T.C.; Olsen, R.J. Lung Cancer Biomarkers: Present Status and Future Developments. Arch. Pathol. Lab. Med. 2013, 137, 1191–1198.
- Cagle, P.T.; Allen, T.C.; Olsen, R.J. Lung Cancer Biomarkers: Present Status and Future Developments. Arch. Pathol. Lab. Med. 2013, 137, 1191–1198.Alberg, A.J.; Brock, M.V.; Ford, J.G.; Samet, J.M.; Spivack, S.D. Epidemiology of Lung Cancer. Chest 2013, 143, e1S–e29S.
- Alberg, A.J.; Brock, M.V.; Ford, J.G.; Samet, J.M.; Spivack, S.D. Epidemiology of Lung Cancer. Chest 2013, 143, e1S–e29S.Sun, S.; Schiller, J.H.; Gazdar, A.F. Lung cancer in never smokers-a different disease. Nat. Rev. Cancer 2007, 7, 778–790.
- Sun, S.; Schiller, J.H.; Gazdar, A.F. Lung cancer in never smokers-a different disease. Nat. Rev. Cancer 2007, 7, 778–790.Herbst, R.S.; Morgensztern, D.; Boshoff, C. The biology and management of non-small cell lung cancer. Nature 2018, 553, 446–454.
- Herbst, R.S.; Morgensztern, D.; Boshoff, C. The biology and management of non-small cell lung cancer. Nature 2018, 553, 446–454.Treatment Choices for Non-Small Cell Lung Cancer, by Stage. Available online: https://www.cancer.org/cancer/lung-cancer/treating-non-small-cell/by-stage.html (accessed on 19 December 2021).
- Treatment Choices for Non-Small Cell Lung Cancer, by Stage. Available online: https://www.cancer.org/cancer/lung-cancer/treating-non-small-cell/by-stage.html (accessed on 19 December 2021).Fennell, D.A.; Summers, Y.; Cadranel, J.; Benepal, T.; Christoph, D.C.; Lal, R.; Das, M.; Maxwell, F.; Visseren-Grul, C.; Ferry, D. Cisplatin in the modern era: The backbone of first-line chemotherapy for non-small cell lung cancer. Cancer Treat. Rev. 2016, 44, 42–50.
- Fennell, D.A.; Summers, Y.; Cadranel, J.; Benepal, T.; Christoph, D.C.; Lal, R.; Das, M.; Maxwell, F.; Visseren-Grul, C.; Ferry, D. Cisplatin in the modern era: The backbone of first-line chemotherapy for non-small cell lung cancer. Cancer Treat. Rev. 2016, 44, 42–50.Li, A.R.; Chitale, D.; Riely, G.J.; Pao, W.; Miller, V.A.; Zakowski, M.F.; Rusch, V.; Kris, M.G.; Ladanyi, M. EGFR mutations in lung adenocarcinomas: Clinical testing experience and relationship to EGFR gene copy number and immunohistochemical expression. J. Mol. Diagn. 2008, 10, 242–248.
- Li, A.R.; Chitale, D.; Riely, G.J.; Pao, W.; Miller, V.A.; Zakowski, M.F.; Rusch, V.; Kris, M.G.; Ladanyi, M. EGFR mutations in lung adenocarcinomas: Clinical testing experience and relationship to EGFR gene copy number and immunohistochemical expression. J. Mol. Diagn. 2008, 10, 242–248.FDA Approvals in Lung Cancer Treatment. Available online: https://www.lungcancerresearchfoundation.org/research/why-research/treatment-advances/ (accessed on 19 December 2021).
- FDA Approvals in Lung Cancer Treatment. Available online: https://www.lungcancerresearchfoundation.org/research/whyresearch/treatment-advances/ (accessed on 19 December 2021).Targeted Drug Therapy for Non-Small Cell Lung Cancer. Available online: https://www.cancer.org/cancer/lung-cancer/treating-non-small-cell/targeted-therapies.html (accessed on 19 December 2021).
- Targeted Drug Therapy for Non-Small Cell Lung Cancer. Available online: https://www.cancer.org/cancer/lung-cancer/treating-non-small-cell/targeted-therapies.html (accessed on 19 December 2021).Schrank, Z.; Chhabra, G.; Lin, L.; Iderzorig, T.; Osude, C.; Khan, N.; Kuckovic, A.; Singh, S.; Miller, R.J.; Puri, N. Current Molecular-Targeted Therapies in NSCLC and Their Mechanism of Resistance. Cancers 2018, 10, 224.
- Schrank, Z.; Chhabra, G.; Lin, L.; Iderzorig, T.; Osude, C.; Khan, N.; Kuckovic, A.; Singh, S.; Miller, R.J.; Puri, N. Current Molecular-Targeted Therapies in NSCLC and Their Mechanism of Resistance. Cancers 2018, 10, 224.Ma, L.-R.; Li, J.-X.; Tang, L.; Li, R.-Z.; Yang, J.-S.; Sun, A.; Leung, E.; Yan, P.-Y. Immune checkpoints and immunotherapy in non-small cell lung cancer: Novel study progression, challenges and solutions (Review). Oncol. Lett. 2021, 22, 1–11.
- Ma, L.-R.; Li, J.-X.; Tang, L.; Li, R.-Z.; Yang, J.-S.; Sun, A.; Leung, E.; Yan, P.-Y. Immune checkpoints and immunotherapy in non-small cell lung cancer: Novel study progression, challenges and solutions (Review). Oncol. Lett. 2021, 22, 1–11.Berghmans, T.; Durieux, V.; Hendriks, L.E.L.; Dingemans, A.-M. Immunotherapy: From Advanced NSCLC to Early Stages, an Evolving Concept. Front. Med. 2020, 7, 90.
- Berghmans, T.; Durieux, V.; Hendriks, L.E.L.; Dingemans, A.-M. Immunotherapy: From Advanced NSCLC to Early Stages, an Evolving Concept. Front. Med. 2020, 7, 90.Walsh, R.J.; Soo, R.A. Resistance to immune checkpoint inhibitors in non-small cell lung cancer: Biomarkers and therapeutic strategies. Ther. Adv. Med. Oncol. 2020, 12, 1758835920937902.
- Walsh, R.J.; Soo, R.A. Resistance to immune checkpoint inhibitors in non-small cell lung cancer: Biomarkers and therapeutic strategies. Ther. Adv. Med. Oncol. 2020, 12, 1758835920937902.Hellmann, M.D.; Ciuleanu, T.E.; Pluzanski, A.; Lee, J.S.; Otterson, G.A.; Audigier-Valette, C.; Minenza, E.; Linardou, H.; Burgers, S.; Salman, P.; et al. Nivolumab plus Ipilimumab in Lung Cancer with a High Tumor Mutational Burden. N. Engl. J. Med. 2018, 378, 2093–2104.
- Hellmann, M.D.; Ciuleanu, T.E.; Pluzanski, A.; Lee, J.S.; Otterson, G.A.; Audigier-Valette, C.; Minenza, E.; Linardou, H.; Burgers, S.; Salman, P.; et al. Nivolumab plus Ipilimumab in Lung Cancer with a High Tumor Mutational Burden. N. Engl. J. Med. 2018, 378, 2093–2104.Gandhi, L.; Rodríguez-Abreu, D.; Gadgeel, S.; Esteban, E.; Felip, E.; De Angelis, F.; Domine, M.; Clingan, P.; Hochmair, M.J.; Powell, S.F.; et al. Pembrolizumab plus Chemotherapy in Metastatic Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2018, 378, 2078–2092.
- Gandhi, L.; Rodríguez-Abreu, D.; Gadgeel, S.; Esteban, E.; Felip, E.; De Angelis, F.; Domine, M.; Clingan, P.; Hochmair, M.J.; Powell, S.F.; et al. Pembrolizumab plus Chemotherapy in Metastatic Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2018, 378, 2078–2092.Kiessling, R.; Klein, E.; Pross, H.; Wigzell, H. “Natural” killer cells in the mouse. II. Cytotoxic cells with specificity for mouse Moloney leukemia cells. Characteristics of the killer cell. Eur. J. Immunol. 1975, 5, 117–121.
- Ahluwalia, P.; Ahluwalia, M.; Mondal, A.K.; Sahajpal, N.S.; Kota, V.; Rojiani, M.V.; Kolhe, R. Natural Killer Cells and Dendritic Cells: Expanding Clinical Relevance in the Non-Small Cell Lung Cancer (NSCLC) Tumor Microenvironment. Cancers 2021, 13, 4037.Chiossone, L.; Dumas, P.-Y.; Vienne, M.; Vivier, E. Natural killer cells and other innate lymphoid cells in cancer. Nat. Rev. Immunol. 2018, 18, 671–688.
- Kiessling, R.; Klein, E.; Pross, H.; Wigzell, H. “Natural” killer cells in the mouse. II. Cytotoxic cells with specificity for mouse Moloney leukemia cells. Characteristics of the killer cell. Eur. J. Immunol. 1975, 5, 117–121.Aribi, M. Introductory Chapter: A Brief Overview on Natural Killer Cells. In Natural Killer Cells; IntechOpen: London, UK, 2017; pp. 1–13.
- Chiossone, L.; Dumas, P.-Y.; Vienne, M.; Vivier, E. Natural killer cells and other innate lymphoid cells in cancer. Nat. Rev. Immunol. 2018, 18, 671–688.Cheent, K.; Khakoo, S.I. Natural killer cells: Integrating diversity with function. Immunology 2009, 126, 449–457.
- Aribi, M. Introductory Chapter: A Brief Overview on Natural Killer Cells. In Natural Killer Cells; IntechOpen: London, UK, 2017; pp. 1–13.Hanna, J.; Bechtel, P.; Zhai, Y.; Youssef, F.; McLachlan, K.; Mandelboim, O. Novel Insights on Human NK Cells’ Immunological Modalities Revealed by Gene Expression Profiling. J. Immunol. 2004, 173, 6547–6563.
- Cheent, K.; Khakoo, S.I. Natural killer cells: Integrating diversity with function. Immunology 2009, 126, 449–457.Vivier, E.; Tomasello, E.; Baratin, M.; Walzer, T.; Ugolini, S. Functions of natural killer cells. Nat. Immunol. 2008, 9, 503–510.
- Hanna, J.; Bechtel, P.; Zhai, Y.; Youssef, F.; McLachlan, K.; Mandelboim, O. Novel Insights on Human NK Cells’ Immunological Modalities Revealed by Gene Expression Profiling. J. Immunol. 2004, 173, 6547–6563.Hu, Z.; Xu, X.; Wei, H. The Adverse Impact of Tumor Microenvironment on NK-Cell. Front. Immunol. 2021, 12, 633361.
- Vivier, E.; Tomasello, E.; Baratin, M.; Walzer, T.; Ugolini, S. Functions of natural killer cells. Nat. Immunol. 2008, 9, 503–510.Cooper, M.A.; Fehniger, T.A.; Caligiuri, M.A. The biology of human natural killer-cell subsets. Trends Immunol. 2001, 22, 633–640.
- Hu, Z.; Xu, X.; Wei, H. The Adverse Impact of Tumor Microenvironment on NK-Cell. Front. Immunol. 2021, 12, 633361.Caligiuri, M.A. Human natural killer cells. Blood 2008, 112, 461–469.
- Cooper, M.A.; Fehniger, T.A.; Caligiuri, M.A. The biology of human natural killer-cell subsets. Trends Immunol. 2001, 22, 633–640.Lanier, L.L.; Le, A.M.; Civin, C.I.; Loken, M.R.; Phillips, J.H. The relationship of CD16 (Leu-11) and Leu-19 (NKH-1) antigen expression on human peripheral blood NK cells and cytotoxic T lymphocytes. J. Immunol. 1986, 136, 4480–4486.
- Caligiuri, M.A. Human natural killer cells. Blood 2008, 112, 461–469.Ferlazzo, G.; Thomas, D.; Lin, S.L.; Goodman, K.; Morandi, B.; Muller, W.A.; Moretta, A.; Münz, C. The abundant NK cells in human secondary lymphoid tissues require activation to express killer cell Ig-like receptors and become cytolytic. J. Immunol. 2004, 172, 1455–1462.
- Lanier, L.L.; Le, A.M.; Civin, C.I.; Loken, M.R.; Phillips, J.H. The relationship of CD16 (Leu-11) and Leu-19 (NKH-1) antigen expression on human peripheral blood NK cells and cytotoxic T lymphocytes. J. Immunol. 1986, 136, 4480–4486.Du, N.; Guo, F.; Wang, Y.; Cui, J. NK Cell Therapy: A Rising Star in Cancer Treatment. Cancers 2021, 13, 4129.
- Ferlazzo, G.; Thomas, D.; Lin, S.L.; Goodman, K.; Morandi, B.; Muller, W.A.; Moretta, A.; Münz, C. The abundant NK cells in human secondary lymphoid tissues require activation to express killer cell Ig-like receptors and become cytolytic. J. Immunol. 2004, 172, 1455–1462.Kärre, K.; Ljunggren, H.G.; Piontek, G.; Kiessling, R. Selective rejection of H-2-deficient lymphoma variants suggests alternative immune defence strategy. Nature 1986, 319, 675–678.
- Du, N.; Guo, F.; Wang, Y.; Cui, J. NK Cell Therapy: A Rising Star in Cancer Treatment. Cancers 2021, 13, 4129.Sivori, S.; Vacca, P.; Del Zotto, G.; Munari, E.; Mingari, M.C.; Moretta, L. Human NK cells: Surface receptors, inhibitory checkpoints, and translational applications. Cell. Mol. Immunol. 2019, 16, 430–441.
- Kärre, K.; Ljunggren, H.G.; Piontek, G.; Kiessling, R. Selective rejection of H-2-deficient lymphoma variants suggests alternative immune defence strategy. Nature 1986, 319, 675–678.Chauvin, J.-M.; Zarour, H.M. TIGIT in cancer immunotherapy. J. Immunother. Cancer 2020, 8, e000957.
- Sivori, S.; Vacca, P.; Del Zotto, G.; Munari, E.; Mingari, M.C.; Moretta, L. Human NK cells: Surface receptors, inhibitory checkpoints, and translational applications. Cell. Mol. Immunol. 2019, 16, 430–441. [CrossRef]Wolf, Y.; Anderson, A.C.; Kuchroo, V.K. TIM3 comes of age as an inhibitory receptor. Nat. Rev. Immunol. 2020, 20, 173–185.
- Chauvin, J.-M.; Zarour, H.M. TIGIT in cancer immunotherapy. J. Immunother. Cancer 2020, 8, e000957.Dai, X.; Zhang, J.; Arfuso, F.; Chinnathambi, A.; Zayed, M.E.; Alharbi, S.A.; Kumar, A.P.; Ahn, K.S.; Sethi, G. Targeting TNF-related apoptosis-inducing ligand (TRAIL) receptor by natural products as a potential therapeutic approach for cancer therapy. Exp. Biol. Med. 2015, 240, 760–773.
- Wolf, Y.; Anderson, A.C.; Kuchroo, V.K. TIM3 comes of age as an inhibitory receptor. Nat. Rev. Immunol. 2020, 20, 173–185.Dong, Y.; Sun, Q.; Zhang, X. PD-1 and its ligands are important immune checkpoints in cancer. Oncotarget 2017, 8, 2171–2186.
- Dai, X.; Zhang, J.; Arfuso, F.; Chinnathambi, A.; Zayed, M.E.; Alharbi, S.A.; Kumar, A.P.; Ahn, K.S.; Sethi, G. Targeting TNFrelated apoptosis-inducing ligand (TRAIL) receptor by natural products as a potential therapeutic approach for cancer therapy. Exp. Biol. Med. 2015, 240, 760–773.Lee, N.; Llano, M.; Carretero, M.; Ishitani, A.; Navarro, F.; López-Botet, M.; Geraghty, D.E. HLA-E is a major ligand for the natural killer inhibitory receptor CD94/NKG2A. Proc. Natl. Acad. Sci. USA 1998, 95, 5199–5204.
- Dong, Y.; Sun, Q.; Zhang, X. PD-1 and its ligands are important immune checkpoints in cancer. Oncotarget 2017, 8, 2171–2186.Barrow, A.D.; Martin, C.J.; Colonna, M. The Natural Cytotoxicity Receptors in Health and Disease. Front. Immunol. 2019, 10, 909.
- Lee, N.; Llano, M.; Carretero, M.; Ishitani, A.; Navarro, F.; López-Botet, M.; Geraghty, D.E. HLA-E is a major ligand for the natural killer inhibitory receptor CD94/NKG2A. Proc. Natl. Acad. Sci. USA 1998, 95, 5199–5204.Pende, D.; Parolini, S.; Pessino, A.; Sivori, S.; Augugliaro, R.; Morelli, L.; Marcenaro, E.; Accame, L.; Malaspina, A.; Biassoni, R.; et al. Identification and molecular characterization of NKp30, a novel triggering receptor involved in natural cytotoxicity mediated by human natural killer cells. J. Exp. Med. 1999, 190, 1505–1516.
- Barrow, A.D.; Martin, C.J.; Colonna, M. The Natural Cytotoxicity Receptors in Health and Disease. Front. Immunol. 2019, 10, 909.Vitale, M.; Bottino, C.; Sivori, S.; Sanseverino, L.; Castriconi, R.; Marcenaro, E.; Augugliaro, R.; Moretta, L.; Moretta, A. NKp44, a novel triggering surface molecule specifically expressed by activated natural killer cells, is involved in non-major histocompatibility complex-restricted tumor cell lysis. J. Exp. Med. 1998, 187, 2065–2072.
- Pende, D.; Parolini, S.; Pessino, A.; Sivori, S.; Augugliaro, R.; Morelli, L.; Marcenaro, E.; Accame, L.; Malaspina, A.; Biassoni, R.; et al. Identification and molecular characterization of NKp30, a novel triggering receptor involved in natural cytotoxicity mediated by human natural killer cells. J. Exp. Med. 1999, 190, 1505–1516.Pessino, A.; Sivori, S.; Bottino, C.; Malaspina, A.; Morelli, L.; Moretta, L.; Biassoni, R.; Moretta, A. Molecular cloning of NKp46: A novel member of the immunoglobulin superfamily involved in triggering of natural cytotoxicity. J. Exp. Med. 1998, 188, 953–960.
- Vitale, M.; Bottino, C.; Sivori, S.; Sanseverino, L.; Castriconi, R.; Marcenaro, E.; Augugliaro, R.; Moretta, L.; Moretta, A. NKp44, a novel triggering surface molecule specifically expressed by activated natural killer cells, is involved in non-major histocompatibility complex-restricted tumor cell lysis. J. Exp. Med. 1998, 187, 2065–2072.Morandi, B.; Bougras, G.; Muller, W.A.; Ferlazzo, G.; Münz, C. NK cells of human secondary lymphoid tissues enhance T cell polarization via IFN-gamma secretion. Eur. J. Immunol. 2006, 36, 2394–2400.
- Pessino, A.; Sivori, S.; Bottino, C.; Malaspina, A.; Morelli, L.; Moretta, L.; Biassoni, R.; Moretta, A. Molecular cloning of NKp46: A novel member of the immunoglobulin superfamily involved in triggering of natural cytotoxicity. J. Exp. Med. 1998, 188, 953–960.Martín-Fontecha, A.; Thomsen, L.L.; Brett, S.; Gerard, C.; Lipp, M.; Lanzavecchia, A.; Sallusto, F. Induced recruitment of NK cells to lymph nodes provides IFN-gamma for T(H)1 priming. Nat. Immunol. 2004, 5, 1260–1265.
- Morandi, B.; Bougras, G.; Muller, W.A.; Ferlazzo, G.; Münz, C. NK cells of human secondary lymphoid tissues enhance T cell polarization via IFN-gamma secretion. Eur. J. Immunol. 2006, 36, 2394–2400.Takeda, K.; Dennert, G. The development of autoimmunity in C57BL/6 lpr mice correlates with the disappearance of natural killer type 1-positive cells: Evidence for their suppressive action on bone marrow stem cell proliferation, B cell immunoglobulin secretion, and autoimmune symptoms. J. Exp. Med. 1993, 177, 155–164.
- Martín-Fontecha, A.; Thomsen, L.L.; Brett, S.; Gerard, C.; Lipp, M.; Lanzavecchia, A.; Sallusto, F. Induced recruitment of NK cells to lymph nodes provides IFN-gamma for T(H)1 priming. Nat. Immunol. 2004, 5, 1260–1265.Lu, L.; Ikizawa, K.; Hu, D.; Werneck, M.B.; Wucherpfennig, K.W.; Cantor, H. Regulation of activated CD4+ T cells by NK cells via the Qa-1–NKG2A inhibitory pathway. Immunity 2007, 26, 593–604.
- Takeda, K.; Dennert, G. The development of autoimmunity in C57BL/6 lpr mice correlates with the disappearance of natural killer type 1-positive cells: Evidence for their suppressive action on bone marrow stem cell proliferation, B cell immunoglobulin secretion, and autoimmune symptoms. J. Exp. Med. 1993, 177, 155–164.Hamilton, G.; Plangger, A. The Impact of NK Cell-Based Therapeutics for the Treatment of Lung Cancer for Biologics: Targets and Therapy. Biologics 2021, 15, 265–277.
- Lu, L.; Ikizawa, K.; Hu, D.; Werneck, M.B.; Wucherpfennig, K.W.; Cantor, H. Regulation of activated CD4+ T cells by NK cells via the Qa-1–NKG2A inhibitory pathway. Immunity 2007, 26, 593–604.Robinson, B.W.; Pinkston, P.; Crystal, R.G. Natural killer cells are present in the normal human lung but are functionally impotent. J. Clin. Investig. 1984, 74, 942–950.
- Hamilton, G.; Plangger, A. The Impact of NK Cell-Based Therapeutics for the Treatment of Lung Cancer for Biologics: Targets and Therapy. Biologics 2021, 15, 265–277.Jabrane-Ferrat, N.; Siewiera, J. The up side of decidual natural killer cells: New developments in immunology of pregnancy. Immunology 2014, 141, 490–497.
- Robinson, B.W.; Pinkston, P.; Crystal, R.G. Natural killer cells are present in the normal human lung but are functionally impotent. J. Clin. Investig. 1984, 74, 942–950.Hanna, J.; Goldman-Wohl, D.; Hamani, Y.; Avraham, I.; Greenfield, C.; Natanson-Yaron, S.; Prus, D.; Cohen-Daniel, L.; Arnon, T.I.; Manaster, I.; et al. Decidual NK cells regulate key developmental processes at the human fetal-maternal interface. Nat. Med. 2006, 12, 1065–1074.
- Jabrane-Ferrat, N.; Siewiera, J. The up side of decidual natural killer cells: New developments in immunology of pregnancy. Immunology 2014, 141, 490–497.Bruno, A.; Focaccetti, C.; Pagani, A.; Imperatori, A.S.; Spagnoletti, M.; Rotolo, N.; Cantelmo, A.R.; Franzi, F.; Capella, C.; Ferlazzo, G.; et al. The proangiogenic phenotype of natural killer cells in patients with non-small cell lung cancer. Neoplasia 2013, 15, 133–142.
- Hanna, J.; Goldman-Wohl, D.; Hamani, Y.; Avraham, I.; Greenfield, C.; Natanson-Yaron, S.; Prus, D.; Cohen-Daniel, L.; Arnon, T.I.; Manaster, I.; et al. Decidual NK cells regulate key developmental processes at the human fetal-maternal interface. Nat. Med. 2006, 12, 1065–1074.Nersesian, S.; Schwartz, S.L.; Grantham, S.R.; MacLean, L.K.; Lee, S.N.; Pugh-Toole, M.; Boudreau, J.E. NK cell infiltration is associated with improved overall survival in solid cancers: A systematic review and meta-analysis. Transl. Oncol. 2021, 14, 100930.
- Bruno, A.; Focaccetti, C.; Pagani, A.; Imperatori, A.S.; Spagnoletti, M.; Rotolo, N.; Cantelmo, A.R.; Franzi, F.; Capella, C.; Ferlazzo, G.; et al. The proangiogenic phenotype of natural killer cells in patients with non-small cell lung cancer. Neoplasia 2013, 15, 133–142.Zhang, S.; Liu, W.; Hu, B.; Wang, P.; Lv, X.; Chen, S.; Shao, Z. Prognostic Significance of Tumor-Infiltrating Natural Killer Cells in Solid Tumors: A Systematic Review and Meta-Analysis. Front. Immunol. 2020, 11, 1242.
- Nersesian, S.; Schwartz, S.L.; Grantham, S.R.; MacLean, L.K.; Lee, S.N.; Pugh-Toole, M.; Boudreau, J.E. NK cell infiltration is associated with improved overall survival in solid cancers: A systematic review and meta-analysis. Transl. Oncol. 2021, 14, 100930.Platonova, S.; Cherfils-Vicini, J.; Damotte, D.; Crozet, L.; Vieillard, V.; Validire, P.; André, P.; Dieu-Nosjean, M.C.; Alifano, M.; Régnard, J.F.; et al. Profound coordinated alterations of intratumoral NK cell phenotype and function in lung carcinoma. Cancer Res. 2011, 71, 5412–5422.
- Zhang, S.; Liu, W.; Hu, B.; Wang, P.; Lv, X.; Chen, S.; Shao, Z. Prognostic Significance of Tumor-Infiltrating Natural Killer Cells in Solid Tumors: A Systematic Review and Meta-Analysis. Front. Immunol. 2020, 11, 1242.Melaiu, O.; Lucarini, V.; Cifaldi, L.; Fruci, D. Influence of the Tumor Microenvironment on NK Cell Function in Solid Tumors. Front. Immunol. 2020, 10.
- Platonova, S.; Cherfils-Vicini, J.; Damotte, D.; Crozet, L.; Vieillard, V.; Validire, P.; André, P.; Dieu-Nosjean, M.C.; Alifano, M.; Régnard, J.F.; et al. Profound coordinated alterations of intratumoral NK cell phenotype and function in lung carcinoma. Cancer Res. 2011, 71, 5412–5422.Masoud, G.N.; Li, W. HIF-1α pathway: Role, regulation and intervention for cancer therapy. Acta Pharm. Sin. B 2015, 5, 378–389.
- Melaiu, O.; Lucarini, V.; Cifaldi, L.; Fruci, D. Influence of the Tumor Microenvironment on NK Cell Function in Solid Tumors. Front. Immunol. 2020, 10.Radomska-Leśniewska, D.M.; Białoszewska, A.; Kamiński, P. Angiogenic Properties of NK Cells in Cancer and Other Angiogenesis-Dependent Diseases. Cells 2021, 10, 1621.
- Masoud, G.N.; Li, W. HIF-1α pathway: Role, regulation and intervention for cancer therapy. Acta Pharm. Sin. B 2015, 5, 378–389.Cerdeira, A.S.; Rajakumar, A.; Royle, C.M.; Lo, A.; Husain, Z.; Thadhani, R.I.; Sukhatme, V.P.; Karumanchi, S.A.; Kopcow, H.D. Conversion of peripheral blood NK cells to a decidual NK-like phenotype by a cocktail of defined factors. J. Immunol. 2013, 190, 3939–3948.
- Radomska-Le´sniewska, D.M.; Białoszewska, A.; Kami ´nski, P. Angiogenic Properties of NK Cells in Cancer and Other Angiogenesis-Dependent Diseases. Cells 2021, 10, 1621.Sadik, S.; Lu, Y.; Zhu, S.; Cai, J.; Mi, L.L. Group 2 innate lymphoid cells (ILC2s): The spotlight in asthma pathogenesis and lung tissue injury. Allergol. Immunopathol. 2021, 49, 208–216.
- Cerdeira, A.S.; Rajakumar, A.; Royle, C.M.; Lo, A.; Husain, Z.; Thadhani, R.I.; Sukhatme, V.P.; Karumanchi, S.A.; Kopcow, H.D. Conversion of peripheral blood NK cells to a decidual NK-like phenotype by a cocktail of defined factors. J. Immunol. 2013, 190, 3939–3948.Shen, C.; Liu, C.; Zhang, Z.; Ping, Y.; Shao, J.; Tian, Y.; Yu, W.; Qin, G.; Liu, S.; Wang, L.; et al. PD-1 Affects the Immunosuppressive Function of Group 2 Innate Lymphoid Cells in Human Non-Small Cell Lung Cancer. Front. Immunol. 2021, 12, 680055.
- Sadik, S.; Lu, Y.; Zhu, S.; Cai, J.; Mi, L.L. Group 2 innate lymphoid cells (ILC2s): The spotlight in asthma pathogenesis and lung tissue injury. Allergol. Immunopathol. 2021, 49, 208–216.Maggi, E.; Veneziani, I.; Moretta, L.; Cosmi, L.; Annunziato, F. Group 2 Innate Lymphoid Cells: A Double-Edged Sword in Cancer? Cancers 2020, 12, 3452.
- Shen, C.; Liu, C.; Zhang, Z.; Ping, Y.; Shao, J.; Tian, Y.; Yu, W.; Qin, G.; Liu, S.; Wang, L.; et al. PD-1 Affects the Immunosuppressive Function of Group 2 Innate Lymphoid Cells in Human Non-Small Cell Lung Cancer. Front. Immunol. 2021, 12, 680055.Jovanovic, I.P.; Pejnovic, N.N.; Radosavljevic, G.D.; Pantic, J.M.; Milovanovic, M.Z.; Arsenijevic, N.N.; Lukic, M.L. Interleukin-33/ST2 axis promotes breast cancer growth and metastases by facilitating intratumoral accumulation of immunosuppressive and innate lymphoid cells. Int. J. Cancer 2014, 134, 1669–1682.
- Maggi, E.; Veneziani, I.; Moretta, L.; Cosmi, L.; Annunziato, F. Group 2 Innate Lymphoid Cells: A Double-Edged Sword in Cancer? Cancers 2020, 12, 3452.Hodge, G.; Barnawi, J.; Jurisevic, C.; Moffat, D.; Holmes, M.; Reynolds, P.N.; Jersmann, H.; Hodge, S. Lung cancer is associated with decreased expression of perforin, granzyme B and interferon (IFN)-γ by infiltrating lung tissue T cells, natural killer (NK) T-like and NK cells. Clin. Exp. Immunol. 2014, 178, 79–85.
- Jovanovic, I.P.; Pejnovic, N.N.; Radosavljevic, G.D.; Pantic, J.M.; Milovanovic, M.Z.; Arsenijevic, N.N.; Lukic, M.L. Interleukin33/ST2 axis promotes breast cancer growth and metastases by facilitating intratumoral accumulation of immunosuppressive and innate lymphoid cells. Int. J. Cancer 2014, 134, 1669–1682.Patel, S.; Vetale, S.; Teli, P.; Mistry, R.; Chiplunkar, S. IL-10 production in non-small cell lung carcinoma patients is regulated by ERK, P38 and COX-2. J. Cell. Mol. Med. 2012, 16, 531–544.
- Hodge, G.; Barnawi, J.; Jurisevic, C.; Moffat, D.; Holmes, M.; Reynolds, P.N.; Jersmann, H.; Hodge, S. Lung cancer is associated with decreased expression of perforin, granzyme B and interferon (IFN)-γ by infiltrating lung tissue T cells, natural killer (NK) T-like and NK cells. Clin. Exp. Immunol. 2014, 178, 79–85.Hatanaka, H.; Abe, Y.; Naruke, M.; Tokunaga, T.; Oshika, Y.; Kawakami, T.; Osada, H.; Nagata, J.; Kamochi, J.-i.; Tsuchida, T.; et al. Significant Correlation between Interleukin 10 Expression and Vascularization through Angiopoietin/TIE2 Networks in Non-small Cell Lung Cancer. Clin. Cancer Res. 2001, 7, 1287–1292.
- Patel, S.; Vetale, S.; Teli, P.; Mistry, R.; Chiplunkar, S. IL-10 production in non-small cell lung carcinoma patients is regulated by ERK, P38 and COX-2. J. Cell. Mol. Med. 2012, 16, 531–544.Neuner, A.; Schindel, M.; Wildenberg, U.; Muley, T.; Lahm, H.; Fischer, J.R. Prognostic significance of cytokine modulation in non-small cell lung cancer. Int. J. Cancer 2002, 101, 287–292.
- Hatanaka, H.; Abe, Y.; Naruke, M.; Tokunaga, T.; Oshika, Y.; Kawakami, T.; Osada, H.; Nagata, J.; Kamochi, J.-i.; Tsuchida, T.; et al. Significant Correlation between Interleukin 10 Expression and Vascularization through Angiopoietin/TIE2 Networks in Nonsmall Cell Lung Cancer. Clin. Cancer Res. 2001, 7, 1287–1292.Wang, Z.; Guan, D.; Huo, J.; Biswas, S.K.; Huang, Y.; Yang, Y.; Xu, S.; Lam, K.-P. IL-10 Enhances Human Natural Killer Cell Effector Functions via Metabolic Reprogramming Regulated by mTORC1 Signaling. Front. Immunol. 2021, 12, 619195.
- Neuner, A.; Schindel, M.; Wildenberg, U.; Muley, T.; Lahm, H.; Fischer, J.R. Prognostic significance of cytokine modulation in non-small cell lung cancer. Int. J. Cancer 2002, 101, 287–292.Castriconi, R.; Cantoni, C.; Della Chiesa, M.; Vitale, M.; Marcenaro, E.; Conte, R.; Biassoni, R.; Bottino, C.; Moretta, L.; Moretta, A. Transforming growth factor beta 1 inhibits expression of NKp30 and NKG2D receptors: Consequences for the NK-mediated killing of dendritic cells. Proc. Natl. Acad. Sci. USA 2003, 100, 4120–4125.
- Wang, Z.; Guan, D.; Huo, J.; Biswas, S.K.; Huang, Y.; Yang, Y.; Xu, S.; Lam, K.-P. IL-10 Enhances Human Natural Killer Cell Effector Functions via Metabolic Reprogramming Regulated by mTORC1 Signaling. Front. Immunol. 2021, 12, 619195.Ferlazzo, G.; Morandi, B. Cross-Talks between Natural Killer Cells and Distinct Subsets of Dendritic Cells. Front. Immunol. 2014, 5, 159.
- Castriconi, R.; Cantoni, C.; Della Chiesa, M.; Vitale, M.; Marcenaro, E.; Conte, R.; Biassoni, R.; Bottino, C.; Moretta, L.; Moretta, A. Transforming growth factor beta 1 inhibits expression of NKp30 and NKG2D receptors: Consequences for the NK-mediated killing of dendritic cells. Proc. Natl. Acad. Sci. USA 2003, 100, 4120–4125.Walzer, T.; Dalod, M.; Robbins, S.H.; Zitvogel, L.; Vivier, E. Natural-killer cells and dendritic cells: “l’union fait la force”. Blood 2005, 106, 2252–2258.
- Ferlazzo, G.; Morandi, B. Cross-Talks between Natural Killer Cells and Distinct Subsets of Dendritic Cells. Front. Immunol. 2014, 5, 159.Adam, C.; King, S.; Allgeier, T.; Braumüller, H.; Lüking, C.; Mysliwietz, J.; Kriegeskorte, A.; Busch, D.H.; Röcken, M.; Mocikat, R. DC-NK cell cross talk as a novel CD4+ T-cell-independent pathway for antitumor CTL induction. Blood 2005, 106, 338–344.
- Walzer, T.; Dalod, M.; Robbins, S.H.; Zitvogel, L.; Vivier, E. Natural-killer cells and dendritic cells: “l’union fait la force”. Blood 2005, 106, 2252–2258.Cong, J.; Wei, H. Natural Killer Cells in the Lungs. Front. Immunol. 2019, 10, 1416.
- Adam, C.; King, S.; Allgeier, T.; Braumüller, H.; Lüking, C.; Mysliwietz, J.; Kriegeskorte, A.; Busch, D.H.; Röcken, M.; Mocikat, R. DC-NK cell cross talk as a novel CD4+ T-cell-independent pathway for antitumor CTL induction. Blood 2005, 106, 338–344.Cong, J.; Wang, X.; Zheng, X.; Wang, D.; Fu, B.; Sun, R.; Tian, Z.; Wei, H. Dysfunction of natural killer cells by FBP1-induced inhibition of glycolysis during lung cancer progression. Cell Metab. 2018, 28, 243–255.e245.
- Cong, J.; Wei, H. Natural Killer Cells in the Lungs. Front. Immunol. 2019, 10, 1416.Della Chiesa, M.; Carlomagno, S.; Frumento, G.; Balsamo, M.; Cantoni, C.; Conte, R.; Moretta, L.; Moretta, A.; Vitale, M. The tryptophan catabolite L-kynurenine inhibits the surface expression of NKp46- and NKG2D-activating receptors and regulates NK-cell function. Blood 2006, 108, 4118–4125.
- Cong, J.; Wang, X.; Zheng, X.; Wang, D.; Fu, B.; Sun, R.; Tian, Z.; Wei, H. Dysfunction of natural killer cells by FBP1-induced inhibition of glycolysis during lung cancer progression. Cell Metab. 2018, 28, 243–255.e245.Mian, M.F.; Lauzon, N.M.; Stämpfli, M.R.; Mossman, K.L.; Ashkar, A.A. Impairment of human NK cell cytotoxic activity and cytokine release by cigarette smoke. J. Leukoc. Biol. 2008, 83, 774–784.
- Della Chiesa, M.; Carlomagno, S.; Frumento, G.; Balsamo, M.; Cantoni, C.; Conte, R.; Moretta, L.; Moretta, A.; Vitale, M. The tryptophan catabolite L-kynurenine inhibits the surface expression of NKp46- and NKG2D-activating receptors and regulates NK-cell function. Blood 2006, 108, 4118–4125.Mian, M.F.; Pek, E.A.; Mossman, K.L.; Stämpfli, M.R.; Ashkar, A.A. Exposure to cigarette smoke suppresses IL-15 generation and its regulatory NK cell functions in poly I:C-augmented human PBMCs. Mol. Immunol. 2009, 46, 3108–3116.
- Mian, M.F.; Lauzon, N.M.; Stämpfli, M.R.; Mossman, K.L.; Ashkar, A.A. Impairment of human NK cell cytotoxic activity and cytokine release by cigarette smoke. J. Leukoc. Biol. 2008, 83, 774–784.Bi, J.; Tian, Z. NK Cell Exhaustion. Front. Immunol. 2017, 8, 760.
- Mian, M.F.; Pek, E.A.; Mossman, K.L.; Stämpfli, M.R.; Ashkar, A.A. Exposure to cigarette smoke suppresses IL-15 generation and its regulatory NK cell functions in poly I:C-augmented human PBMCs. Mol. Immunol. 2009, 46, 3108–3116.Li, Q.; Cai, S.; Li, M.; Zhou, X.; Wu, G.; Kang, K.; Yuan, J.; Wang, R.; Huyan, T.; Zhang, W. Natural killer cell exhaustion in lung cancer. Int. Immunopharmacol. 2021, 96, 107764.
- Bi, J.; Tian, Z. NK Cell Exhaustion. Front. Immunol. 2017, 8, 760.
More