Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Apurba Chakrabarti and Version 2 by Catherine Yang.
Mitral valve prolapse (MVP) is a common cause of valvular heart disease. Although many patients with MVP have a benign course, there is increasing recognition of an arrhythmic phenotype associated with ventricular arrhythmias and sudden cardiac death (SCD). Pathophysiologic mechanisms associated with arrhythmias include cardiac fibrosis, mechanical stress induced changes in ventricular refractory periods, as well as electrophysiologic changes in Purkinje fibers.
- mitral valve prolapse
- sudden cardiac death
1. Introduction
Mitral valve prolapse (MVP) is the abnormal systolic excursion of mitral valve leaflets into the left atrium. First described clinically in the 1960s with cine-angiocardiography [1], it is now defined as at least 2 mm of superior displacement of mitral valve leaflets above the mitral annulus, measured echocardiographically in the parasternal or apical long axis views. Using modern definitions, MVP has a prevalence between 1 and 3% [2][3][4][2,3,4]. MVP is among the most common causes of chronic primary mitral regurgitation, although only a minority of patients will progress to severe mitral regurgitation [5].
Pathologically, MVP is a spectrum of anatomical abnormalities ranging from the classic form (Barlow’s Disease) with thickened redundant leaflets to a non-classic form (fibroelastic deficiency) with thinner leaflets [6][7][6,7]. In classic MVP, myxoid infiltration results in leaflet thickening ≥ 5 mm, bileaflet prolapse with redundant and billowing leaflets, mitral annular dilation, and chordal changes. In contrast, non-classic MVP is associated with leaflet thinning due to reduced connective tissue production and more often has segmental prolapse [7]. MVP can be a primary abnormality or secondary to another syndrome. Primary MVP is typically sporadic, but there are familial/genetic patterns. Several genes (FLNA, DCHS1, DZIP1, and PLD1) with a wide variety of inheritance patterns have been associated with familial MVP and techniques such as exome sequencing and genome-wide association studies are identifying novel genes as well [7][8][9][7,8,9]. FLNC mutations are associated with an arrhythmogenic form of MVP [10]. Alternatively, MVP can be secondary to connective tissue syndromes (e.g., Marfan, Ehlers–Danlos, Loeys–Dietz) or a variety of other cardiac disorders.
MVP is related to but distinct from mitral annular disjunction (MAD). The mitral annulus is an anatomically “D”-shaped structure, into which the anterior and posterior mitral valve leaflets insert [11][12][11,12]. MAD is defined as mitral annular detachment from the basal left ventricular (LV) myocardium with an abnormal systolic excursion of the leaflet hinge point into the left atrium. Pathologically, MAD was first described in the context of MVP in the 1980s, but MAD has been observed in the absence of MVP [13][14][15][16][13,14,15,16]. Abnormal systolic curling of the posterior mitral annulus, which was first described in MVP patients in the 1970s, has recently been shown to strongly correlate with MAD [17][18][17,18]. The displacement distance can be measured non-invasively by echocardiography, cardiac computed tomography (CT), and cardiac magnetic resonance (CMR). In subsequent studies of various cohorts of MVP, MAD has been identified in approximately 15–55% of patients with significant variation based on the different cohorts studied and imaging modalities utilized [19][20][21][22][23][24][25][26][27][19,20,21,22,23,24,25,26,27]. A systematic review found MAD in about 33% of MVP patients [19].
The long-term outcome of patients with MVP is heterogenous [2][5][28][2,5,28]. While many patients do well, there is increasing recognition of a subgroup of patients with an arrhythmic course involving ventricular arrhythmias (VA) and sudden cardiac death (SCD) [4][29][30][31][4,29,30,31]. Given the relatively high prevalence of MVP, the identification and management of MVP patients at risk of SCD is critical.
2. Arrhythmogenesis
While poorly understood, the current understanding of arrhythmogenesis in MVP involves the development of a substrate for arrhythmias (cardiac fibrosis) combined with a trigger for arrhythmias (Figure 1). The anatomic defect of MVP, leaflet prolapse, causes abnormal tension on the papillary muscles and abnormal wall stress on adjacent LV basal and inferolateral myocardium. Long-term mechanical stress may result in inflammation or localized ischemia leading to replacement fibrosis. Both histologic and MRI studies have confirmed the presence of fibrosis in these areas [30]. Furthermore, a recent hybrid PET/MRI study found FDG uptake (evidence of inflammation or ischemia) and cardiac fibrosis in these areas (and others) [32].
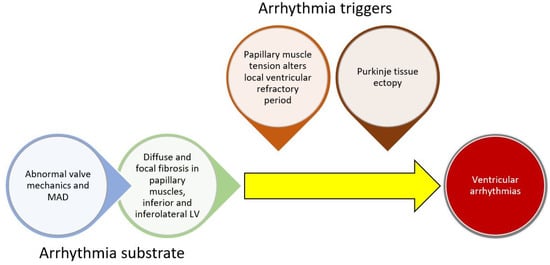
Figure 1. Conceptual framework for ventricular arrhythmogenesis involving the development of cardiac substrate and triggers for arrhythmias.
In addition to a substrate, a trigger is typically necessary for sustained VA. Two key theories for triggers involve abnormal mechanical stretch and Purkinje tissue. In animal models, papillary muscle traction prolonged the ventricular functional refractory period locally [33]. Given the abnormal mechanics of prolapsing leaflets in MVP, local variation in the refractory period may induce or sustain papillary muscle arrhythmias. In addition, Purkinje fibers arborize in areas adjacent to papillary muscle and in the inferior/inferolateral myocardium. They may also trigger VA in MVP patients. In one study of MVP patients undergoing ablation, all patients who had a cardiac arrest had a Purkinje origin of their arrhythmias [34]. Another study found that Purkinje potentials at the site of ablation were associated with successful ablation [34][35][34,35]. This has also been observed in a broader array of non-MVP patients. In a study of patients with papillary muscle PVCs undergoing ablation, Purkinje potentials preceded the clinical PVCs in over half of the patients [36]. It is possible that abnormal mechanical tension induces changes in the electrophysiologic function in papillary muscles and possibly the Purkinje fibers and acts as a trigger for VA in areas of cardiac fibrosis. Furthermore, the mitral regurgitation itself may lead to cardiac remodeling and further triggers for arrhythmias.
MAD has also been associated with VA both in the presence and absence of MVP. This has led some authors to suggest that MAD is the inciting factor in the cascade that involves altered valvular mechanics, valvular degeneration, leaflet prolapse, myocardial fibrosis, and finally arrhythmias. This has been labeled the Padua hypothesis [37], and while promising, requires further research to confirm causal links.
3. Natural History
The majority of patients with MVP have a benign course. However, there are subgroups at higher risk for complications. Studies are difficult to generalize due to a variety of cohorts studied. In a large study of Olmstead Countypatients, those with asymptomatic MVP had a 10-year cardiovascular morbidity and overall mortality of 30% and 19%, respectively [38]. Another study showed a similar mortality of 13% at 8 years [39]. At least half of mortality and morbidity events were felt to be related to MVP directly [38]. Aside from mortality, complications of MVP include the need for cardiac surgery, heart failure, atrial fibrillation, stroke, arterial thromboembolism, and endocarditis [28][38][40][28,38,40]. After 5.4 years of median follow-up, 7.8% of patients required mitral valve surgery [38].
Sudden cardiac death (SCD) and VA occur at a low rate in patients with MVP, about 0.14 per 100 patient years in community-dwelling patients [4]. Some cohorts have reported higher rates of SCD of 0.2–0.4% or higher, but these studies may have bias from referral to tertiary centers and higher-risk clinical factors [28][40][41][42][28,40,41,42]. With 8 years of follow-up, about 17% patients required ICD or VT ablation in one study of MVP patients referred to a tertiary center [39]. Although the absolute rate of SCD is low overall, the relatively high prevalence of MVP translates to a significant population at risk of SCD. MVP has been observed in about 2% of all SCD cases [4][43][4,43], but may explain as high as 10–12% in younger patients with SCD or patients with unexplained SCD [4][44][4,44].