Idiopathic pulmonary fibrosis (IPF) is a fibrosing interstitial lung disease of unknown etiology. Different types of cells are involved in fibrogenesis, which is persistently physical and molecular stimulation, either directly or by interacting with bioactive molecules and extracellular vesicles (EVs). Current evidence suggests that EVs play an essential role in IPF development. EVs are released by a variety of cells, including fibroblasts, epithelial cells, and alveolar macrophages. In addition, EVs can transport bioactive molecules, such as lipids, proteins, and nucleic acids, which play a pivotal role in cellular communication. Several proposed mechanisms show that an acceptor cell can capture, absorb, or interact with EVs through direct fusion with the plasma membrane, ligand–receptor interaction, and endocytotic process, modifying the target cell. During fibrogenesis, the release of EVs is deregulated, increases the EVs amount, and the cargo content is modified. This alteration is closely associated with the maintenance of the fibrotic microenvironment.
- extracellular vesicles
- fibroblasts
- macrophages
- alveolar epithelial cells
- mesenchymal stem cells
- therapeutic
1. Idiopathic Pulmonary Fibrosis
2. EVs and Their Involvement in Cellular and Molecular Mechanisms Associated with IPF
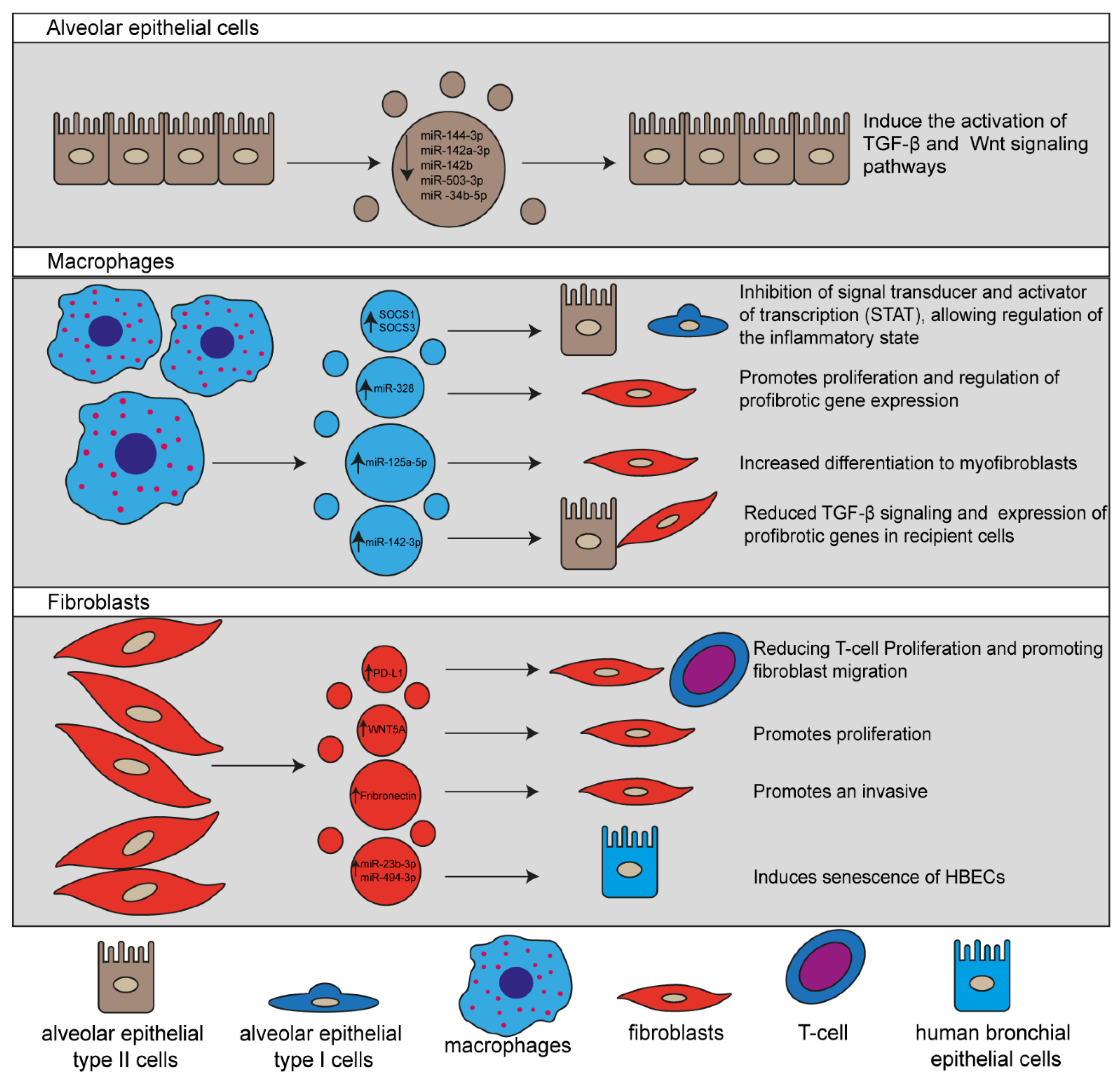 Figure 1. Schematic representation of EVs released by different lung cells involved in Idiopathic pulmonary fibrosis (IPF) pathogenesis. EVs and their cargo molecules regulate the main signaling pathways associated with profibrotic processes facilitating the IPF progression.
Figure 1. Schematic representation of EVs released by different lung cells involved in Idiopathic pulmonary fibrosis (IPF) pathogenesis. EVs and their cargo molecules regulate the main signaling pathways associated with profibrotic processes facilitating the IPF progression.
2.1. Cells Involved in IPF Development
The involvement of cells in the fibrogenesis of IPF has been extensively studied; nevertheless, the exact mechanism of EVs in promoting cellular communication leading to IPF development and evolution remains unclear [4][5]. Current knowledge on the pathogenesis of IPF has demonstrated that during the early stage of the disease, chronic tissue injury promote apoptosis of type I alveolar epithelial cell (AEC-I), recruitment of inflammatory cells, mainly macrophages and neutrophils, activation of type II alveolar epithelial cell (AEC-II) that regenerate damaged cells, and the release of proinflammatory cytokines that promote the recruitment, activation, and proliferation of fibroblasts [6][4]. Thus, fibroblasts are the most important cells in IPF development since they undergo differentiation into myofibroblasts facilitating their capability to produce and secrete ECM components [4].2.1.1. EVs Released by Alveolar Epithelial Cells
A critical step during IPF development is the persistent injury of AEC-I and II by environmental factors. During this process, AECs may undergo apoptosis, release inflammatory mediators. EVs that strongly contribute to IPF development [4][7]. Recently, it has been reported that syndecan-1 protein is increased in both lung epithelium of IPF patients and murine bleomycin-induced IPF models. Likewise, authors also demonstrated that syndecan-1 plays an essential role in the packaging of antifibrotic miRNAs in EVs, including miR-144-3p, miR-142a-3p, miR-142b, miR-503-3p, and miR-34b-5p, which have shown diverse effects on signaling pathways associated with fibroproliferation and fibrogenesis, suggesting that those miRNAs play a pivotal role in the regulation of IPF progress. Moreover, they observed that by incubating AEC-II with EVs isolated from wild-type and Sdc1−/− mice models of IPF induced with bleomycin, EVs from wild-type mice act as a profibrotic signal promoting the activation of different signaling pathways involved in fibrotic processes, such as TGF-β and Wnt as compared with the effect of EVs isolated from Sdc1−/− mice [7].2.1.2. EVs Secreted by Macrophages
Alveolar macrophages secrete EVs to transport the suppressor of cytokine signaling 1 and 3 (SOCS1 and SOCS3, respectively). Consequently, they are primarily taken up by AECs, resulting in the inhibition of signal transducer and activator of transcription (STAT), which regulates the inflammatory response, indicating that EVs derived from macrophages are involved in the pulmonary homeostasis2.1.3. EVs Released by Fibroblasts
Fibroblasts are in charge of restoring the ECM components after injury; nonetheless, in the pathogenesis of IPF, this capability is altered by the persistent exposition to profibrotic factors, which leads to increased production of ECM components3. Involvement of MSCs in Lung Repair
Previous studies have proposed that mesenchymal stem cells (MSCs) are responsible for maintaining vascular homeostasis and facilitating repair; contrarily, studies performed in lung tissue have reported that MSCs participates in the restoration of injured endothelium through the release of paracrine mediators, such as EVs and activation of different signaling pathways, including (IL-6), interleukin 6 receptor subunit alpha (IL-6RA), Janus kinase (JAK), signal transducer and activator of transcription 3 (STAT3) . In vitro studies demonstrated that human lung resident MSCs (Lr-MSCs) can decrease fibroblasts proliferation and increase the ability to induce wound closure in alveolar epithelial cells A5493.1. EVs Released by MSCs as Therapeutic Mediators
3.1. EVs Released by MSCs as Therapeutic Mediators
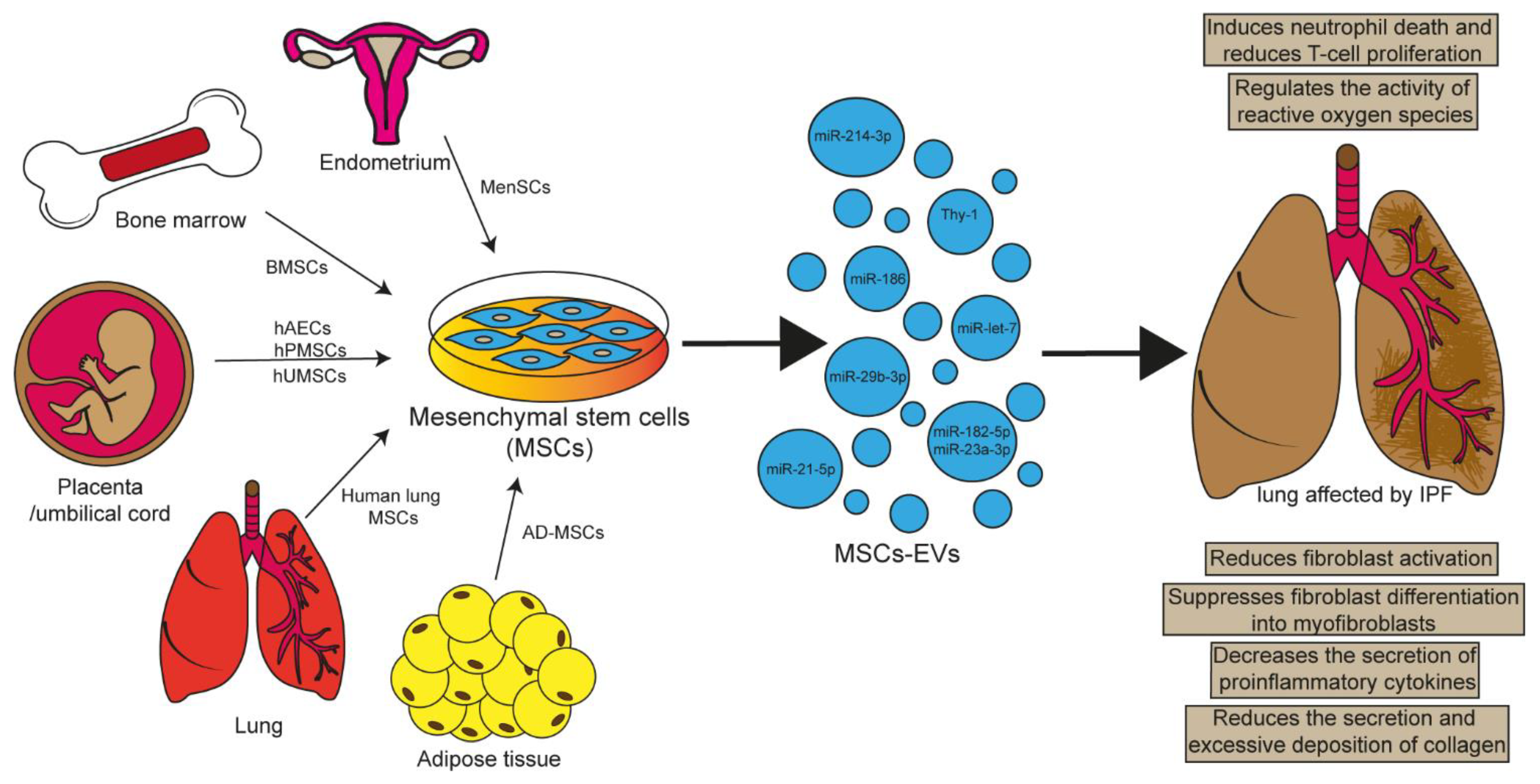 Figure 2.
Figure 2.| Experimental Model | EVs Source | Cargo | Effects | Reference |
|---|
| EVs Source | Cargo | Target | Effect | Reference |
|---|
| Radiation-induced lung injury | hP-MSCs | miR-214-3p | Attenuates pulmonary vascular damage, inflammation, and fibrosis. | [24] |
| Bleomycin-induced IPF | BMSCs | miR-186 | Relieves IPF by blocking fibroblasts activation by suppressing SOX4 and DKK1 expression. | [28] |
| E. coli endotoxin induced ALI | BMSCs | KGF mRNA | Restores lung protein permeability and reduces inflammation. | [27] |
| IRI-induced ALI | BMSCs | miR-21-5p | Decreases edema, pulmonary dysfunction, M1 polarization of alveolar macrophages, and secretion of the cytokines IL-8, IL-1β, IL-6, IL-17, and TNF-α. | [29] |
| Hypoxia-induced PH | BMSCs | unknown | Prevents activation of hypoxic signaling, lung inflammation, and PH development through inhibition of hypoxic STAT3 signaling. | [30] |
| Bleomycin-induced IPF | BMSCs | miR-29b-3p |
| hAECs | unknown | HLF activated with TGF-β | Inhibit fibroblasts activation | [35] | |||||
| BMSCs | miR-186 | Fibroblasts LL29 |
Inhibit fibroblasts activation by supression of SOX4 and DKK1 | [27] | |||||
| BMSCs | interaction of Thy-1 with beta integrins | CCL-210 (HLF) | Blocks myofbroblastic diferentiation | [37] | |||||
| BMSCs | miR-29b-3p | LL29 | Inhibit the activation of fibroblasts through FZD6 | [31]BMSCs | miR-21-5p | ] | |||
| Dendritic cells activated with LPS |
Increased production of antiinflammatory cytokines Reduced synthesis of proinflammatory cytokines, such as IL-6 and IL-12 Decreased migratory capacity |
[38] | Attenuates IPF progression by suppressing fibroblasts proliferation, migration, and differentiation through suppression of FZD6 expression. | [31] | |||||
| BMSCs | miR-182-5p and miR-23a-3p, | MLE-12 cell activated with LPS | Inactivate NF-κB and hedgehog pathways Reverts EMT |
[32] | LPS-induced ALI | BMSCs | miR-23a-3p y miR-182-5p | Attenuate lung injury, EMT, and fibrosis by inhibiting NF-κB and hedgehog pathways. | |
| Human lung MSCs | unknown | IB3-1 cells activated with TNF-α |
Attenuate the expresión of inflammatory cytokines Increase antioxidant intrinsic defenses | [32 | [39] | LPS-induced ALI | AD-MSCs | unknown | They reduce inflammation, alveolar septal thickening, alter macrophage phenotypes, reduce levels of the proinflammatory cytokine IL-1β, and increase anti-inflammatory IL-10. |
| MenSCs | miR-Let-7 | MLE-12 cells activated with BLM | [ | Inhibits pulmonary fibrosis through regulation of ROS, mtDNA damage and NLRP3 inflammasome activation. | 33] | ||||
| [ | 36 | ] | LPS-induced ALI | AD-MSCs | miR-27a-3p | Alleviates lung injury, inhibits NF-κB activation and promotes M2 polarization of alveolar macrophages. | [34] | ||
| Bleomycin-induced IPF | |||||||||
| AD-MSCs | unknown | Macrophage activated with LPS | Suppressed the activation of proinflammatory genes IL-6, IL-1β, TNF-α | [33hAECs | unknown | Attenuates inflammation and pulmonary fibrosis. | [35] | ||
| Bleomycin-induced IPF | MenSCs | miR-Let-7 | Attenuates lung inflammation and fibrosis by regulating ROS, mtDNA damage, and NLRP3 inflammasome activation. | [36] |
| ] | ||
| AD-MSCs | ||
| miR-27a-3p | ||
| Macrophage activated with LPS | Inhibits NF-κB activation and promotes M2 polarization. | [34] |
| EVs Source | Cargo | Target | Effect | Reference |
|---|---|---|---|---|
| hAECs | unknown | HLF activated with TGF-β | Inhibit fibroblasts activation | [35] |
| BMSCs | miR-186 | Fibroblasts LL29 |
Inhibit fibroblasts activation by supression of SOX4 and DKK1 | [27] |
| BMSCs | interaction of Thy-1 with beta integrins | CCL-210 (HLF) | Blocks myofbroblastic diferentiation | [37] |
| BMSCs | miR-29b-3p | LL29 | Inhibit the activation of fibroblasts through FZD6 | [31] |
| BMSCs | miR-21-5p | Dendritic cells activated with LPS |
Increased production of antiinflammatory cytokines Reduced synthesis of proinflammatory cytokines, such as IL-6 and IL-12 Decreased migratory capacity |
[38] |
| BMSCs | miR-182-5p and miR-23a-3p, | MLE-12 cell activated with LPS | Inactivate NF-κB and hedgehog pathways Reverts EMT |
[32] |
| Human lung MSCs | unknown | IB3-1 cells activated with TNF-α |
Attenuate the expresión of inflammatory cytokines Increase antioxidant intrinsic defenses |
[39] |
| MenSCs | miR-Let-7 | MLE-12 cells activated with BLM |
Inhibits pulmonary fibrosis through regulation of ROS, mtDNA damage and NLRP3 inflammasome activation. | [36] |
| AD-MSCs | unknown | Macrophage activated with LPS | Suppressed the activation of proinflammatory genes IL-6, IL-1β, TNF-α | [33] |
| AD-MSCs | miR-27a-3p | Macrophage activated with LPS | Inhibits NF-κB activation and promotes M2 polarization. | [34] |
3.1.1. hUMSC-EVs
3.1.1. hUMSC-EVs
3.1.2. hAEC-EVs
3.1.2. hAEC-EVs
3.1.3. AD-MSCs-EVs
3.1.3. AD-MSCs-EVs
3.1.4. BMSC-EVs
3.1.4. BMSC-EVs
3.1.5. MenSC-EVs
3.1.5. MenSC-EVs
4. Conclusions and Perspectives
In conclusion, IPF is a chronic progressive pathology of unknown etiology, poor prognosis, and high mortality rate. This disease involves a heterogeneous group of cells such as AEC-I, AEC-II, macrophages, and fibroblasts that orchestrate a pathological state mediated by a microenvironment enriched with soluble factors associated with fibrosis development, such as cytokines, chemokines, and EVs production. In recent years, it has been shown that EVs participate in the initiation, and progression of the pathophysiological mechanisms associated with IPF; for example, immunomodulation, fibroproliferation, differentiation, and mesenchymal–epithelial transition through the transfer of bioactive cargo molecules, such as proteins (PD-L1, WNT5A and Fibronectin) and miRNAs (miR-328, miR-125a-5p, miR-142-3p, miR-23b-3p and miR-494-3p). Thus, EVs can modulate the functioning of the host cells, accelerating lung damage and leading to decreased gas exchange by excessive ECM deposition. Therefore, these crucial features strongly suggest that EVs play an essential role in the pathogenesis of IPF and may be useful for achieving a better understanding of the disease and identifying candidates for biomarkers for the diagnosis, prognosis of IPF. However, there are still many challenges ahead; despite the number of available studies describing how EVs and their cargo molecules play an essential role in IPF progression, the mechanism by which these EVs regulate cellular processes involved in IPF development remains to be fully elucidated. Therefore, further exploration and new studies are needed to expand the understanding of EVs and their cargo molecules in the pathogenesis of IPF.References
- Richeldi, L.; Collard, H.R.; Jones, M.G. Idiopathic pulmonary fibrosis. Lancet 2017, 389, 1941–1952.
- Lederer, D.J.; Longo, D.L.; Martinez, F.J. Idiopathic Pulmonary Fibrosis. N. Engl. J. Med. 2018, 378, 1811–1823.
- Martin-Medina, A.; Lehmann, M.; Burgy, O.; Hermann, S.; Baarsma, H.A.; Wagner, D.E.; De Santis, M.M.; Ciolek, F.; Hofer, T.P.; Frankenberger, M.; et al. Increased Extracellular Vesicles Mediate WNT5A Signaling in Idiopathic Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2018, 198, 1527–1538.
- Sgalla, G.; Iovene, B.; Calvello, M.; Ori, M.; Varone, F.; Richeldi, L. Idiopathic pulmonary fibrosis: Pathogenesis and management. Respir. Res. 2018, 19, 1–18.
- Kubo, H. Extracellular Vesicles in Lung Disease. Chest 2018, 153, 210–216.
- Martinez, F.J.; Collard, H.R.; Pardo, A.; Raghu, G.; Richeldi, L.; Selman, M.; Swigris, J.J.; Taniguchi, H.; Wells, A.U. Idiopathic pulmonary fibrosis. Nat. Rev. Dis. Primers 2017, 3, 1–19.
- Parimon, T.; Yao, C.; Habiel, D.M.; Ge, L.; Bora, S.A.; Brauer, R.; Evans, C.M.; Xie, T.; Alonso-Valenteen, F.; Medina-Kauwe, L.K.; et al. Syndecan-1 promotes lung fibrosis by regulating epithelial reprogramming through extracellular vesicles. JCI Insight 2019, 4.
- Peters-Golden, M.; Curtis, J.L.; Freeman, C.M.; Mancuso, P.; Swanson, J.A.; Przybranowski, S.; Schneider, D.J.; Speth, J.M.; Penke, L.R.K.; Zasłona, Z.; et al. Transcellular delivery of vesicular SOCS proteins from macrophages to epithelial cells blunts inflammatory signaling. J. Exp. Med. 2015, 212, 729–742.
- Yao, M.-Y.; Zhang, W.-H.; Ma, W.-T.; Liu, Q.-H.; Xing, L.-H.; Zhao, G.-F. microRNA-328 in exosomes derived from M2 macrophages exerts a promotive effect on the progression of pulmonary fibrosis via FAM13A in a rat model. Exp. Mol. Med. 2019, 51, 1–16.
- Qin, X.; Lin, X.; Liu, L.; Li, Y.; Li, X.; Deng, Z.; Chen, H.; Chen, H.; Niu, Z.; Li, Z.; et al. Macrophage-derived exosomes mediate silica-induced pulmonary fibrosis by activating fibroblast in an endoplasmic reticulum stress-dependent manner. J. Cell. Mol. Med. 2021, 25, 4466–4477.
- Wang, D.; Hao, C.; Zhang, L.; Zhang, J.; Liu, S.; Li, Y.; Qu, Y.; Zhao, Y.; Huang, R.; Wei, J.; et al. Exosomal miR-125a-5p derived from silica-exposed macrophages induces fibroblast transdifferentiation. Ecotoxicol. Environ. Saf. 2020, 192, 110253.
- Guiot, J.; Cambier, M.; Boeckx, A.; Henket, M.; Nivelles, O.; Gester, F.; Louis, E.; Malaise, M.; Dequiedt, F.; Louis, R.; et al. Macrophage-derived exosomes attenuate fibrosis in airway epithelial cells through delivery of antifibrotic miR-142-3p. Thorax 2020, 75, 870–881.
- Wang, T.; Gilkes, D.M.; Takano, N.; Xiang, L.; Luo, W.; Bishop, C.J.; Chaturvedi, P.; Green, J.J.; Semenza, G.L. Hypoxia-inducible factors and RAB22A mediate formation of microvesicles that stimulate breast cancer invasion and metastasis. Proc. Natl. Acad. Sci. USA 2014, 111, E3234–E3242.
- Chanda, D.; Otoupalova, E.; Hough, K.P.; Locy, M.L.; Bernard, K.; Deshane, J.S.; Sanderson, R.D.; Mobley, J.A.; Thannickal, V.J. Fibronectin on the Surface of Extracellular Vesicles Mediates Fibroblast Invasion. Am. J. Respir. Cell Mol. Biol. 2019, 60, 279–288.
- Kang, J.H.; Jung, M.Y.; Choudhury, M.; Leof, E.B. Transforming growth factor beta induces fibroblasts to express and release the immunomodulatory protein PD-L1 into extracellular vesicles. FASEB J. 2019, 34, 2213–2226.
- Kadota, T.; Yoshioka, Y.; Fujita, Y.; Araya, J.; Minagawa, S.; Hara, H.; Miyamoto, A.; Suzuki, S.; Fujimori, S.; Kohno, T.; et al. Extracellular Vesicles from Fibroblasts Induce Epithelial-Cell Senescence in Pulmonary Fibrosis. Am. J. Respir. Cell Mol. Biol. 2020, 63, 623–636.
- Zhou, Y.; Li, P.; Goodwin, A.J.; Cook, J.A.; Halushka, P.V.; Chang, E.; Fan, H. Exosomes from Endothelial Progenitor Cells Improve the Outcome of a Murine Model of Sepsis. Mol. Ther. 2018, 26, 1375–1384.
- Tadokoro, T.; Wang, Y.; Barak, L.S.; Bai, Y.; Randell, S.H.; Hogan, B.L.M. IL-6/STAT3 promotes regeneration of airway ciliated cells from basal stem cells. Proc. Natl. Acad. Sci. USA 2014, 111, E3641–E3649.
- Shi, W.; Hostettler, K.E.; Gazdhar, A.; Khan, P.; Savic, S.; Tamo, L.; Lardinois, D.; Roth, M.; Tamm, M.; Geiser, T. Multipotent mesenchymal stem cells in lung fibrosis. PLoS ONE 2017, 12, e0191144.
- Jun, D.; Garat, C.; West, J.; Thorn, N.; Chow, K.; Cleaver, T.; Sullivan, T.; Torchia, E.C.; Childs, C.; Shade, T.; et al. The Pathology of Bleomycin-Induced Fibrosis Is Associated with Loss of Resident Lung Mesenchymal Stem Cells That Regulate Effector T-cell Proliferation. Stem Cells 2011, 29, 725–735.
- Xiang, B.; Chen, L.; Wang, X.; Zhao, Y.; Wang, Y.; Xiang, C. Transplantation of Menstrual Blood-Derived Mesenchymal Stem Cells Promotes the Repair of LPS-Induced Acute Lung Injury. Int. J. Mol. Sci. 2017, 18, 689.
- Bacha, N.C.; Blandinieres, A.; Rossi, E.; Gendron, N.; Nevo, N.; Lecourt, S.; Guerin, C.L.; Renard, J.M.; Gaussem, P.; Angles-Cano, E.; et al. Endothelial Microparticles are Associated to Pathogenesis of Idiopathic Pulmonary Fibrosis. Stem Cell Rev. Rep. 2017, 14, 223–235.
- Ionescu, L.; Byrne, R.N.; van Haaften, T.; Vadivel, A.; Alphonse, R.S.; Rey-Parra, G.J.; Weissmann, G.; Hall, A.; Eaton, F.; Thébaud, B. Stem cell conditioned medium improves acute lung injury in mice: In vivo evidence for stem cell paracrine action. Am. J. Physiol. -Lung Cell. Mol. Physiol. 2012, 303, L967–L977.
- Lei, X.; He, N.; Zhu, L.; Zhou, M.; Zhang, K.; Wang, C.; Huang, H.; Chen, S.; Li, Y.; Liu, Q.; et al. Mesenchymal Stem Cell-Derived Extracellular Vesicles Attenuate Radiation-Induced Lung Injury via miRNA-214-3p. Antioxid. Redox Signal. 2021, 35, 849–862.
- Wiklander, O.P.B.; Brennan, M.Á.; Lötvall, J.; Breakefield, X.O.; El Andaloussi, S. Advances in therapeutic applications of extracellular vesicles. Sci. Transl. Med. 2019, 11, eaav8521.
- Kim, S.Y.; Joglekar, M.V.; Hardikar, A.A.; Phan, T.H.; Khanal, D.; Tharkar, P.; Limantoro, C.; Johnson, J.; Kalionis, B.; Chrzanowski, W. Placenta Stem/Stromal Cell–Derived Extracellular Vesicles for Potential Use in Lung Repair. Proteomics 2019, 19, 1800166.
- Zhu, Y.-g.; Feng, X.-m.; Abbott, J.; Fang, X.-h.; Hao, Q.; Monsel, A.; Qu, J.-m.; Matthay, M.A.; Lee, J.W. Human Mesenchymal Stem Cell Microvesicles for Treatment of Escherichia coli Endotoxin-Induced Acute Lung Injury in Mice. Stem Cells 2014, 32, 116–125.
- Zhou, J.; Lin, Y.; Kang, X.; Liu, Z.; Zhang, W.; Xu, F. microRNA-186 in extracellular vesicles from bone marrow mesenchymal stem cells alleviates idiopathic pulmonary fibrosis via interaction with SOX4 and DKK1. Stem Cell Res. Ther. 2021, 12, 1–14.
- Li, J.w.; Wei, L.; Han, Z.; Chen, Z. Mesenchymal stromal cells-derived exosomes alleviate ischemia/reperfusion injury in mouse lung by transporting anti-apoptotic miR-21-5p. Eur. J. Pharmacol. 2019, 852, 68–76.
- Lee, C.; Mitsialis, S.A.; Aslam, M.; Vitali, S.H.; Vergadi, E.; Konstantinou, G.; Sdrimas, K.; Fernandez-Gonzalez, A.; Kourembanas, S. Exosomes Mediate the Cytoprotective Action of Mesenchymal Stromal Cells on Hypoxia-Induced Pulmonary Hypertension. Circulation 2012, 126, 2601–2611.
- Wan, X.; Chen, S.; Fang, Y.; Zuo, W.; Cui, J.; Xie, S. Mesenchymal stem cell-derived extracellular vesicles suppress the fibroblast proliferation by downregulating FZD6 expression in fibroblasts via micrRNA-29b-3p in idiopathic pulmonary fibrosis. J. Cell. Physiol. 2020, 235, 8613–8625.
- Xiao, K.; He, W.; Guan, W.; Hou, F.; Yan, P.; Xu, J.; Zhou, T.; Liu, Y.; Xie, L. Mesenchymal stem cells reverse EMT process through blocking the activation of NF-κB and Hedgehog pathways in LPS-induced acute lung injury. Cell Death Dis. 2020, 11, 1–17.
- Huang, R.; Qin, C.; Wang, J.; Hu, Y.; Zheng, G.; Qiu, G.; Ge, M.; Tao, H.; Shu, Q.; Xu, J. Differential effects of extracellular vesicles from aging and young mesenchymal stem cells in acute lung injury. Aging 2019, 11, 7996–8014.
- Wang, J.; Huang, R.; Xu, Q.; Zheng, G.; Qiu, G.; Ge, M.; Shu, Q.; Xu, J. Mesenchymal Stem Cell–Derived Extracellular Vesicles Alleviate Acute Lung Injury Via Transfer of miR-27a-3p. Crit. Care Med. 2020, 48, e599–e610.
- Tan, J.L.; Lau, S.N.; Leaw, B.; Nguyen, H.P.T.; Salamonsen, L.A.; Saad, M.I.; Chan, S.T.; Zhu, D.; Krause, M.; Kim, C.; et al. Amnion Epithelial Cell-Derived Exosomes Restrict Lung Injury and Enhance Endogenous Lung Repair. Stem Cells Transl. Med. 2018, 7, 180–196.
- Sun, L.; Zhu, M.; Feng, W.; Lin, Y.; Yin, J.; Jin, J.; Wang, Y. Exosomal miRNA Let-7 from Menstrual Blood-Derived Endometrial Stem Cells Alleviates Pulmonary Fibrosis through Regulating Mitochondrial DNA Damage. Oxidative Med. Cell. Longev. 2019, 2019, 1–17.
- Shentu, T.-P.; Huang, T.-S.; Cernelc-Kohan, M.; Chan, J.; Wong, S.S.; Espinoza, C.R.; Tan, C.; Gramaglia, I.; van der Heyde, H.; Chien, S.; et al. Thy-1 dependent uptake of mesenchymal stem cell-derived extracellular vesicles blocks myofibroblastic differentiation. Sci. Rep. 2017, 7, 18052.
- Reis, M.; Mavin, E.; Nicholson, L.; Green, K.; Dickinson, A.M.; Wang, X.-N. Mesenchymal Stromal Cell-Derived Extracellular Vesicles Attenuate Dendritic Cell Maturation and Function. Front. Immunol. 2018, 9, 2538.
- Zulueta, A.; Colombo, M.; Peli, V.; Falleni, M.; Tosi, D.; Ricciardi, M.; Baisi, A.; Bulfamante, G.; Chiaramonte, R.; Caretti, A. Lung mesenchymal stem cells-derived extracellular vesicles attenuate the inflammatory profile of Cystic Fibrosis epithelial cells. Cell. Signal. 2018, 51, 110–118.
- Dong, L.; Wang, Y.; Zheng, T.; Pu, Y.; Ma, Y.; Qi, X.; Zhang, W.; Xue, F.; Shan, Z.; Liu, J.; et al. Hypoxic hUCMSC-derived extracellular vesicles attenuate allergic airway inflammation and airway remodeling in chronic asthma mice. Stem Cell Res. Ther. 2021, 12, 1–14.
- Ridzuan, N.; Zakaria, N.; Widera, D.; Sheard, J.; Morimoto, M.; Kiyokawa, H.; Mohd Isa, S.A.; Chatar Singh, G.K.; Then, K.-Y.; Ooi, G.-C.; et al. Human umbilical cord mesenchymal stem cell-derived extracellular vesicles ameliorate airway inflammation in a rat model of chronic obstructive pulmonary disease (COPD). Stem Cell Res. Ther. 2021, 12, 1–21.
- Mansouri, N.; Willis, G.R.; Fernandez-Gonzalez, A.; Reis, M.; Nassiri, S.; Mitsialis, S.A.; Kourembanas, S. Mesenchymal stromal cell exosomes prevent and revert experimental pulmonary fibrosis through modulation of monocyte phenotypes. JCI Insight 2019, 4.