Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Yasemin van Heuvel and Version 2 by Dean Liu.
The genomic RNA of HIV-1 enables employing complex splicing patterns the encryption of multiple proteins within a limited coding capacity. The viral RNA and related proteins mediate toxicity via multiple different pathways. The circumvention of the host cell immune system and the progressing elimination of T helper cells leads to AIDS and opportunistic infections. The aberrant interaction of viral RNA-binding proteins can result in cellular transcriptional deregulations, tumor formation and apoptosis.
- retroviruses
- mRNA splicing
- cytotoxicity
1. Genome and Virion Structure
The RNA genome (gRNA) of HIV-1, with approximately 9 kb, is considerably small. However, it contains all necessary information to synthesize all 15 proteins needed for replication and assembly of new virions in the infected host cells [1][2][14,15]. The viral genome encapsulated in virions consists of a dimer of single stranded positively sensed gRNAs. The different open reading frames (ORFs) are illustrated in Figure 1, except for the ORF encoding the antisense protein, yet uncharacterized for its role in the replication cycle [1][3][14,16]. The genome encompasses nine different ORFs and some of the viral genes overlap, thus enabling the encryption of many proteins within a limited coding capacity. The genome is flanked by the long terminal repeats (LTRs). They contain the essential information—including the viral promoter—for gene expression, integration, and reverse transcription and are divided into the U3, R, and U5 elements [4][10]. The cis-acting regulatory element U3 is divided into a modulatory, an enhancer, and a basal region and contains three binding sites for splice factors as well as two binding sites for host cell transcription factors, e.g., Nuclear Factor-κB (NF-κB). The R element contains the trans-acting responsive region (TAR), forming a RNA stem-loop structure that plays an important role in viral replication, i.e., the activation of transcription [5][6][17,18]. The U5 element contains the polyadenylation signal (poly A) and regulatory regions for reverse transcription. The U5 element is followed by the primer binding site (PBS), the dimerization initiation signal (DIS), and the major splice-donor site (D1), all not shown in Figure 1. The packaging signal Psi (ψ) mediates the packaging of the viral gRNA [7][19]. The consecutive gag gene encodes the structural viral core proteins. The precursor protein p55-Gag is processed by the viral protease during virion maturation into the subunits matrix (MA), capsid (CA) and nucleocapsid (NC) proteins. The pol gene encodes the subunit viral enzymes protease (PR), reverse transcriptase (RT), and integrase (IN), also originating from a precursor protein upon viral protease-mediated cleavage. The third structural gene env encodes the two envelope glycoproteins gp120-SU (surface unit) and the gp41-TM (transmembrane unit). The pol gene is followed by the two regulatory genes rev and tat as well as four accessory genes vif, vpr, and vpu. Tat and Rev are indispensable for viral replication, accumulate within the host cell nucleus and bind to their cognate mRNA structures, namely, the Rev-responsive element (RRE) and TAR. Rev is an important nuclear export factor that mediates the transport of partially spliced and unspliced viral mRNAs into the cytoplasm. Tat is a strong transcriptional activator [8][9][10][20,21,22]. Vif, Vpr, and Vpu influence the rate of virus particle production. The accessory nef gene at the end of the gRNA elevates HIV infectivity and downregulates several host cell proteins including CD4 and the major histocompatibility complex I (MHC I) [11][23]. Moreover, Vif, Vpu, and Nef counteract several cellular restriction factors to secure efficient replication. Table 1 provides an overview of the best characterized restriction factors.

Figure 1. HIV-1 genome and virion structure. (Top) Schematic overview of the genomic organization of the HIV-1 genome encompassing the open reading frames coding for the different structural, regulatory, and accessory proteins.
The mature membrane-enveloped HIV-1 virion is spherical in shape with a diameter of approximately 120 nm. The virion’s lipid bilayer membrane contains, besides several host cell proteins, ~7–35 envelope trimers consisting of gp120-SU and the gp41-TM [11][12][13][14][23,31,32,33]. Both proteins are encoded in the env gene and originate from the Env polyprotein gp160 upon cleavage by the cellular furin-like protease [15][27]. The membrane envelopes the matrix protein (p17-MA) formed core. The viral capsid is formed by 1000 to 1500 cone-shaped hexameric capsid proteins (p24-CA) [16][34]. The capsid encapsulates two copies of positive-sense and single-stranded gRNAs stabilized by the nucleocapsid proteins (p7-NC). The mature virion harbors the viral enzymes reverse transcriptase (p66-/p51-RT), protease (p10-PR), integrase (p32-IN), and the accessory protein Vpr that are needed in the maturation process [11][17][23,35].
2. Receptors and Cell Entry
Figure 2 provides an overview of the HIV-1 replication cycle. The HIV-1 infection of a host cell is receptor-dependent and begins with the binding of the envelope protein gp120-SU to the primary host cell receptor CD4 and the co-receptors, chemokine receptor type 5 (CCR5), or C-X-C motif chemokine receptor type 4 (CXCR4). The binding induces conformational changes of the envelope protein trimers, which leads to the fusion of the virion with the host cell membrane [18][36]. In more detail, when Env binds to the co-receptor, the virus exposes the fusion peptide at the N-terminus of gp41-TM, which inserts into the cell membrane. Again, dramatic conformational rearrangements, forming a very stable six-helix bundle, pull both membranes into close proximity, reaching a hemifusion state initiating in a last step the fusion of both membranes [18][19][20][36,37,38]. Although cryo-electron microscopic images of this process exist, many structural aspects of the proteins involved are still not fully understood. Once the fusion pore opens, the virion releases its interior into the cytoplasm of the host cell [21][39].
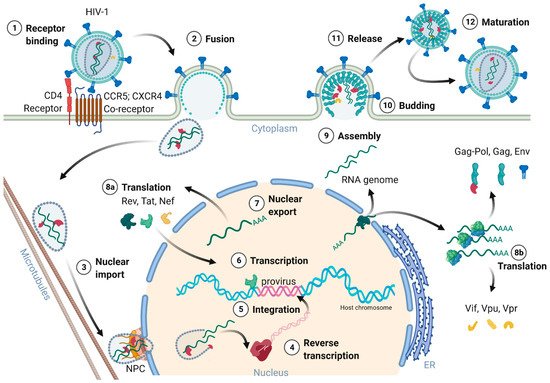
Figure 2. Schematic overview of the HIV-1 replication cycle.
3. Nuclear Entry, Reverse Transcription, and Uncoating
The cone shaped ~60 nm in diameter capsid, consisting of 250 hexamers and 12 pentamers, was believed to partially uncoat or disassemble already within the cytoplasm [8][11][18][22][20,23,36,40]. However, most recent studies of Zila and colleagues in 2021 provided astonishing insights into the viral capsid and its trafficking along the microtubules of the cell towards the nuclear pore complex (NPC), revealing that the entire capsid enters the nucleus [22][40]. As the capsid enters the cytoplasm, it travels along the microtubules towards the nucleus aided by dynein and kinesin-1. Next, the capsid docks with its narrow end to the NPC interacting with the NPC-proteins Nup358 and Nup62. Upon nucleoplasm entry, the capsid partially disassembles, releasing the CA interior [22][23][40,41]. Dharan and colleagues discovered that the uncoating as well as reverse transcription are completed within the host cell nucleus [23][41], which was confirmed by two other studies of Burdick and colleagues [24][42] as well as Müller and co-workers [25][43] showing that proviral DNA could only be detected inside the nucleus. Therefore, the reverse transcription already starts within the intact capsid and is finalized upon capsid nucleus entry [24][25][42,43]. Burdick et al. also discovered that the complete uncoating takes place 1.5 h before provirus integration into the host cell genome and within a range of 1.5 μm proximate to the gene-rich loci in the euchromatin regions.
The reverse transcription of the viral gRNA to proviral dsDNA in infected cells is an important step of the replication cycle. The RNA/DNA-dependent DNA polymerase and RNAse H are part of p66-RT, whereas p51-RT provides conformational stability. The reverse transcription starts with the so-called first strand transfer and the synthesis of the single stranded DNA (ssDNA). The ssDNA is hybridized to the 3′-end of the viral genome and the negative strand DNA synthesis continues. The second strand transfer leads then to the transcription of the positive strand DNA and dsDNA synthesis is finalized [26][44]. Template switching events and error-prone RT activity contribute to the high genetic variability of HIV [27][45].
4. Genome Integration
Retroviruses permanently integrate their reverse transcribed proviruses into the host cell genome, making the virus an everlasting part of the infected host cell. The integrated provirus can remain dormant within the host, and thus escape from the immune system’s detection and response. These properties render HIV to a latent and life-long infection [28][46].
The proviral integration is mediated by the viral IN in concert with RT [4][10]. The integrase forms together with the provirus a strong nucleoprotein complex targeting active transcription units for integration into the genome [29][47]. These units are found in clusters within the less condensed euchromatin characterized by high transcriptional activity. The integration process is divided into two steps. First, the 3′-ends of the provirus is processed and the two terminal nucleotides are removed, exposing a 3′-hydroxyl group and a 5′-overhang. Next, the targeted host DNA is cleaved, and the processed provirus is integrated, ligating the 3′-ends with the 5′-ends of the target DNA [30][31][48,49].
5. Transcription, Splicing, and Protein Expression
After integration of the provirus, it either remains transcriptionally silent and enters latency or initiates the production of new virions. The protein expression of HIV-1 is regulated at the epigenetic, transcriptional, and posttranscriptional level [32][33][34][50,51,52]. Latently infected cells serve as viral reservoirs, resisting eradication during ART and by the immune system due to the absence of target viral protein expression. Latency is induced by infection of resting cells not supporting efficient viral transcription, by inactive proviral integration sites, epigenetic silencing, and by the differentiation of infected effector immune cells to resting memory cells, respectively [34][35][52,53]. However, transcription of the provirus and replication can be reactivated.
The HIV-1 provirus utilizes the host transcription machinery. Host transcription factors such as NF-κB, specificity protein 1 (Sp1) and activator protein 1 (AP-1) are known activators of HIV transcription [32][33][36][50,51,54]. General transcription factors, mediator, and RNA polymerase II (RNA Pol II) assemble into the preinitiation complex at the 5′-LTR promoter. The HIV-1 5′-LTR contains three possible transcription start sites (TSS) consisting of three consecutive guanosins (G) at the junction between the R and U3 region. Depending on the TSS used for transcription, the untranslated 5′-region (5′-UTR) of the proviral RNA transcript begins with a single, two, or three G residues [37][55]. Promoter clearing is mediated by the phosphorylation of the C-terminal domain of RNA pol II mediated by the transcription factor TFIIH [38][39][56,57]. A short RNA segment of about 60 nucleotides is transcribed before promoter-proximal pausing occurs. The pausing is triggered by the formation of the TAR RNA stem-loop and the binding of negative transcription elongation factors (N-TEFs) to the preinitiation complex [6][40][41][18,58,59]. The pause is released by Tat binding to TAR, acting as a transcription factor activating positive transcription elongation factor b kinase (P-TEFb) [6][42][43][18,60,61]. In cells, the majority of P-TEFb is part of the 7SK small nuclear ribonucleoprotein (7SK snRNP), in which the catalytic activity of P-TEFb is inhibited by the Hexim-1 protein [44][62]. McNamara and colleagues suggested a model of Tat-mediated recruitment of the protein phosphatase 1G (PPM1G) to 7SK snRNP to the HIV promoter [43][61]. PPM1G then dephosphorylates P-TEFb, thus releasing it from the 7Sk snRNP complex. When Tat binds to the released P-TEFb it induces re-phosphorylation. Tat and the activated P-TEFb kinase bind to TAR, bringing the kinase in proximity to the stalled RNA Pol II transcription complex. P-TEFb phosphorylates the C-terminal domain of RNA Pol II and N-TEFs, facilitating the elongation of the viral transcript [6][43][45][18,61,63].
The HIV provirus undergoes three transcription phases [35][53]: During latency no virions are produced, although stochastic transcriptional bursts at the LTR promoter occur [46][64]. Upon cell activation, e.g., by immune stimuli, host transcription factors such as NF-κB can reactivate viral transcription and induce the expression of Tat protein, enabling a positive feedback loop. The Tat-mediated transcriptional boost results in the production of full-length gRNA ready to be encapsidated or serving as templates for alternative splicing. The full-length gRNA consists of nine partially overlapping ORFs. Therefore, it is alternatively spliced to generate mRNAs, encoding all viral proteins [1][2][47][14,15,65]. The mRNAs are categorized into three classes: (I) full-length, unspliced ~9 kb gRNA, (II) intron-containing, partially spliced ~4 kb mRNAs, and (III) intronless, fully spliced ~2 kb mRNAs [2][48][15,66]. The gag and pol gene products are translated from the unspliced full-length gRNA, whereas the other viral proteins Nef, Rev, Tat, Env precursor protein, Vpr, Vif, and Vpu are produced from either partially or fully spliced mRNAs. Figure 3 provides an overview on the mRNA classes as well as splice donor and acceptor sites present in the HIV-1 mRNA transcript. All HIV mRNAs that undergo splicing utilize the major splice donor site (D1), which defines the first exon between the 5′-Cap and D1 included in all viral mRNAs [47][48][65,66]. The exon defined by D4 and either the splice acceptors A3, A4, or A5 and the final exon between A7 and the poly A tail are additional constitutive exons present in all HIV mRNAs [48][66]. The full-length gRNA transcript is sequentially spliced, starting at D1 to a downstream splice acceptor site and a prerequisite for further downstream splicing [49][67]. The packaging signal Ψ is removed, and thus ensures selective full-length gRNAs encapsidation into new virions [50][68]. Splicing of the viral mRNAs is tightly regulated by the cellular spliceosome. As the splicing of D1 to a downstream splice acceptor is mandatory for all subsequent splice events, suppression of splicing at D1 results in unspliced transcripts [48][49][66,67]. Noteworthy, the 5′-UTR of the full-length transcript can adopt different secondary conformations depending on the number of guanosines at the 5′-Cap [51][69]. RNAs that start with a 1GCap fold into a structure that masks D1 and favors the formation of RNA dimers, whereas RNAs with 2GCap or 3GCap fold differently and expose the D1 site for splicing [37][52][55,70]. To generate partially spliced mRNAs, splicing events are regulated by a complex interplay of several splicing regulatory elements that modulate the usage of splice sites [2][15]. Unspliced and partially spliced mRNAs harbor the intron, spanning from D4 to A7. This is pivotal as this intron contains the RRE indispensable for the Rev-mediated nuclear export of intron-containing mRNAs.
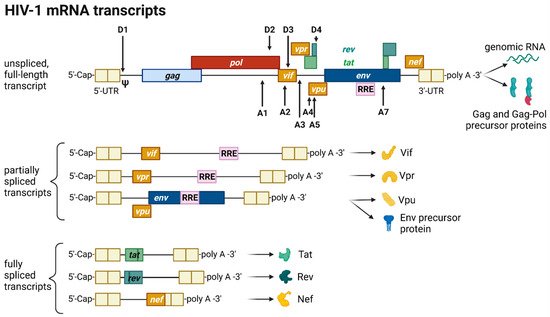
Figure 3. HIV-1 mRNA transcripts and splice sites.
Only intronless mRNAs are exported across the NPC by cellular mRNA export pathways. Consequently, only the fully spliced viral mRNA transcripts are exported to the cytoplasm and translated early in the viral replication cycle, first enabling the expression of Tat, Rev, and Nef proteins. In contrast, incompletely spliced, intron-containing mRNAs are excluded from the nuclear export pathway and degraded [53][54][71,72]. Once expressed, Rev is transported into the nucleus, where it accumulates and co-transcriptionally binds RRE present in incompletely spliced viral transcripts mediating nuclear export [53][55][71,73]. This way, HIV circumvents the nuclear mRNA degradation of RRE-containing transcripts. Rev recruits the cellular export factor chromosomal maintenance 1 (CRM1), which mediates the RanGTP-dependent export of the Rev:RNA:CRM-1 complex to the cytoplasm [32][53][55][50,71,73].
In summary, viral gene expression is regulated via transcription, splicing patterns, and RNA structures. Early in the viral gene expression only fully processed mRNAs are translated into the accessory protein Nef and the regulatory proteins Tat and Rev. Nef increases viral infectivity by remodeling signal pathways, downregulating the expression of cell surface proteins such as CD4, major histocompatibility complex-I, and activation of viral transcription through NF-κB [56][57][74,75]. Tat activates and stimulates transcription of the provirus by interaction with cellular co-factors at the TAR RNA structure. Rev enables the export of RRE-containing incompletely processed RNAs, shifting the viral protein expression to proteins necessary for the production of new virions. The mRNAs encoding the p55-Gag precursor, p160-Gag-Pol precursor, and Vif and Vpr proteins are translated by polysomes in the cytosol [58][76]. The Gag-Pol precursor proteins are translated from the full-length gRNA by a ribosomal frameshift during translation [59][77]. The bicistronic vpu/env mRNA is translated into Vpu and Env precursor gp160 in the rough endoplasmic reticulum (ER). Inside the ER, the Env precursor gp160 assembles into trimers and travels to the Golgi apparatus, in which gp160 gets glycosylated and cleaved by furin-like proteases into the mature Env glycoprotein complex consisting of the subunits gp120-SU and gp41-TM [60][78]. Env and Vpu are transported to the plasma membrane via the secretory pathway for incorporation into assembling viral particles [60][78]. In conclusion, all components needed to initiate virus assembly are now available.
6. Assembly, Budding, and Virion Maturation
The viral structural Gag precursor protein is sufficient for the formation of new particles. Gag consists of four structural domains separated by protease cleavage sites: the N-terminal MA domain, the CA domain, the NC domain flanked by two spacer peptides (SP1 and SP2), and the C-terminal p6 domain. Each domain performs specific functions during assembly and budding of the viral particle via interactions with viral and cellular proteins and RNAs. The gRNA molecules form a dimer selectively recruited for packaging. Intramolecular and intermolecular interactions of gRNA and Gag polyprotein mediate the selective packaging of the viral genome into assembling particles. The 5′-UTR of the gRNA folds into complex structures consisting of several stem-loops, including the packaging signal Ψ and the dimerization initiation signal (DIS). Recent studies by the Summers group revealed that gRNAs exhibiting a sequestered 1GCap at the 5′-UTR are preferentially packaged and adopt a dimer competent conformation [37][61][62][55,79,80]. In this conformation, the DIS is exposed and two gRNA molecules dimerize through intermolecular DIS base pairing. The gRNA dimers expose several binding sites located in the DIS and Ψ stem-loops for the interaction with the NC domain of the Gag precursor proteins [63][81]. Binding of gRNA also promotes the dimerization of Gag by protein-protein interactions [64][65][82,83]. The Gag:gRNA complex travels to and is anchored in the plasma membrane through the N-terminal myristoylation signal present in the MA domain. HIV-1 assembles at the cell membrane in specific cholesterol- and phosphatidylinositol-(4,5)-bisphosphate (PI(4,5)P2)-rich microdomains called lipid rafts. The targeting of Gag to the membrane is regulated by the electrostatical interaction of the highly basic regions located in the MA domain with PI(4,5)P2 and the binding of tRNALys, which prevents binding of MA to intracellular membranes [66][67][68][84,85,86]. In addition and upon simultaneous binding of PI(4,5)P2 and gRNA, Gag folds from a compact to an extended conformation enabling the anchoring of the myristoylation signal to the plasma membrane and initiating the multimerization of Gag proteins [69][70][87,88]. Gag and Gag-Pol protein multimerization at the plasma membrane is stabilized by CA-CA and CA-SP1 protein-protein interactions [71][89]. The assembly of Gag at the plasma membrane also induces the retention of Env trimers at assembly sites mediated by an interaction between the Gag MA domain and the C-tail of the Env protein gp41-TM [60][78]. In addition to Env, the p6 domain of Gag captures Vpr [72][90]. The growing Gag multimer bends the membrane and forms a spherical nascent particle still connected to the membrane. However, and for the release of the particle, HIV-1 relies on the cellular endosomal sorting complexes required for transport (ESCRT) machinery [73][91]. Gag recruits the ESCRT complexes via adaptor proteins, which recognize amongst others the amino acid motifs PTAP and LYPX(n)L present in the p6 domain. Tumor susceptibility gene 101 protein (Tsg101) is part of the ESCRT-I complex, binds to the PTAP motif, and forms a supercomplex with ESCRT-II, whereas the adaptor protein apoptosis-linked gene 2-interacting protein X (Alix) recognizes LYPX(n)L and interacts with ESCRT-III. The ESCRT-III complex constricts the membrane and catalyzes the release of the immature particle [73][91].
The viral particle matures and reorganizes its structural proteins, gRNAs, and enzymes, resulting in the formation of an infectious virion. The maturation is initiated by the auto-activation of the PR sequentially cleaving the Gag and Gag-Pol precursor proteins releasing the viral enzymes PR, RT, and IN and the structural proteins p17-MA, p24-CA, and p7-NC [74][75][92,93]. The structural changes are mandatory for viral infectivity. The NC protein binds tightly to the gRNA dimer and stabilizes linkage between the two gRNA molecules [63][76][81,94]. The CA proteins assemble around the NC:gRNA complex encapsidating the viral genome as well as RT and IN [74][92]. The processing of Gag into its subunits renders the incorporated Env trimers’ fusogenicity. The HIV-1 virion concludes the productive cell infection and is now armed for a new replication cycle [77][95].
7. Cytoxicity of HIV Infection
RNAs are able to cause diseases in many different ways controlling and also disrupting multiple genetic and metabolic pathways in the cell [78][96]. For example, the transcription of non-coding repeat expansions can lead to toxic RNAs—e.g., the dominantly inherited and multisystemic disease myotonic dystrophy type 1 (DM1), where CTG repeat expansions in the 3′UTR of the DM1 protein kinase (DMPK) gene generate DMPK mRNAs that are trapped in ribonuclear foci, compromising the availability of RNA-binding protein (RBP) levels. RNA foci are believed to sequestrate bound RBPs and result in toxicity [79][80][97,98]. Many disease-related genes encode RBPs, where mutated gene products accumulate as aggregates disrupting cellular functions involved in RNA metabolism [81][82][99,100]. Mutations in the RBPs, TAR DNA (TARDBP), FUS RNA-binding protein (FUS), Ataxin 2 (ATXN2) as well as EWS RNA-binding proteins (EWSR1) and many more have been shown to greatly influence disease risks, e.g., amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FDT) [82][100].
RNAs also play a pivotal role in the HIV infection cycle and pathogenesis. Viral gene expression is regulated via transcription splicing patterns and RNA structures interacting with viral and host cell RBPs. Cellular RBPs are strongly recruited away from their cellular functions and cellular cognate target RNAs in response to viral infection, which skews the availability of target RNAs towards HIV transcripts [83][101]. Maybe most importantly, the two viral regulatory trans-acting nuclear RBPs of HIV, Tat and Rev bind cis-acting RNA motifs, the TAR and RRE of the newly transcribed HIV genomic RNA, and thus mediate the deregulation of the host cell RNA and protein synthesis machinery to enable efficient virus replication [84][85][102,103]. As illustrated in Figure 4, TAR (located in the HIV leader RNA element) and RRE (located in the HIV env gene) motifs fold into complex secondary RNA structures folding into highly conserved stem loops and bulges. Rev and RRE are known to assemble to a homo-oligomeric ribonucleoprotein complex needed for the nuclear export of intron containing messenger RNAs from the nucleus into the cytoplasm. RRE as well as TAR are also known as target RNA structures for small molecules intervening the HIV replication cycle. However, until today, little is known about the cytotoxic and disease-causing effects of Rev-RRE in contrast to Tat-TAR [85][86][103,104].
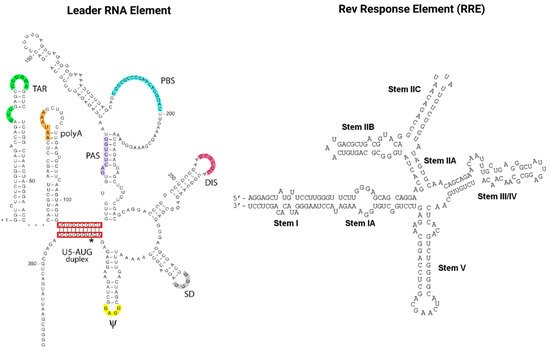
Figure 4. The cis-acting RNA regulatory elements of HIV-1. The untranslated highly conserved leader RNA including TAR (left) and the RRE (right).
Tat recruits the histone acetyltransferases to the viral promoter to activate the transcription of the viral genome. In addition, the RNA helicase A (RHA) acts as a strong TAR-binding cellular co-factor and enhances HIV-1 LTR-driven gene expression and virus production. The RBP Tat enters the nucleus and binds to the host cell RBP P-TEFb. This complex then interacts with TAR on the RNA enhancing the activity of RNA-Pol II, and thus transcription levels [78][87][96,105]. Tat’s role as the trans-activator of HIV transcription is fully characterized. Other replication-independent effects mediated by the viral soluble protein Tat cause diseases. Cells constantly release Tat into the extracellular space where it exerts cytotoxicity harming cells in proximity, also known as bystander toxicity, as illustrated in Figure 5 [86][104].
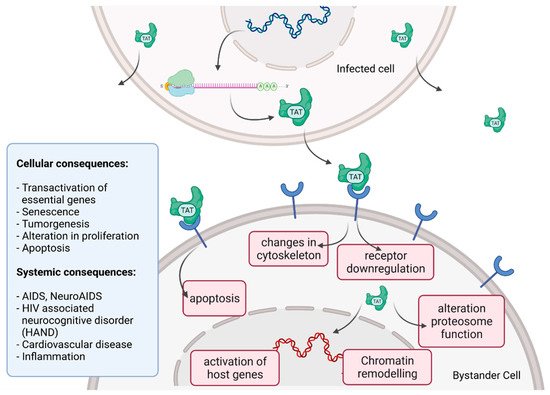
Figure 5.
HIV Tat bystander toxicity.
Upon infection, Tat accumulates at the inside of the plasma membrane of infected cells and is released into the extracellular compartment. Tat actively recruits monocytes and macrophages into the areas of infection. By binding to a variety of cell surface receptors, e.g., heparan sulfate proteoglycans (HSPGs), chemokine receptors, integrins and lipoprotein receptor-related protein-1 (LRP-1), Tat is able to penetrate into a range of different cell types, amongst others, monocytes, macrophages, lymphocytes, astrocytes, neurons and cardiomyocytes. Here, Tat induces the release of mainly pro-inflammatory chemokines and cytokines (e.g., CCL2, TNF-α, IL-2, IL-6, IL-8) that activate transmigration and can be toxic to uninfected bystanding cells as cardiomyocytes and the heart. Tat alters the activity of the proteosome complex (e.g., down regulation of cellular proteins and up regulation of viral proteins). As one example, Tat induces the upregulation of Connexin 43 mRNA and proteins in cardiomyocytes and increases lipofuscin levels, a known aging heart biomarker. Tat also leads to the alteration of actin filaments, tight junctions and adhesion molecules, altering the organization of the cytoskeleton. Inside the nucleus Tat recruits RBPs and binds TAR inducing transcriptional regulation of gene expression and chromatin remodeling resulting in many different cellular and systemic alterations [78][86][96,104]. In the case of HIV-associated neurocognitive disorder (HAND), Tat can induce neurotoxicity directly as well as indirectly by triggering inflammation through the activation and recruitment of macrophages, microglia and astrocytes into the affected areas of the brain [86][104]. Latest findings suggest that Tat causes the emergence of neurocognitive and cardiovascular impairments in about 50 to 60% of HIV-infected individuals as a result of Tat’s bystander toxicity [86][88][104,106].