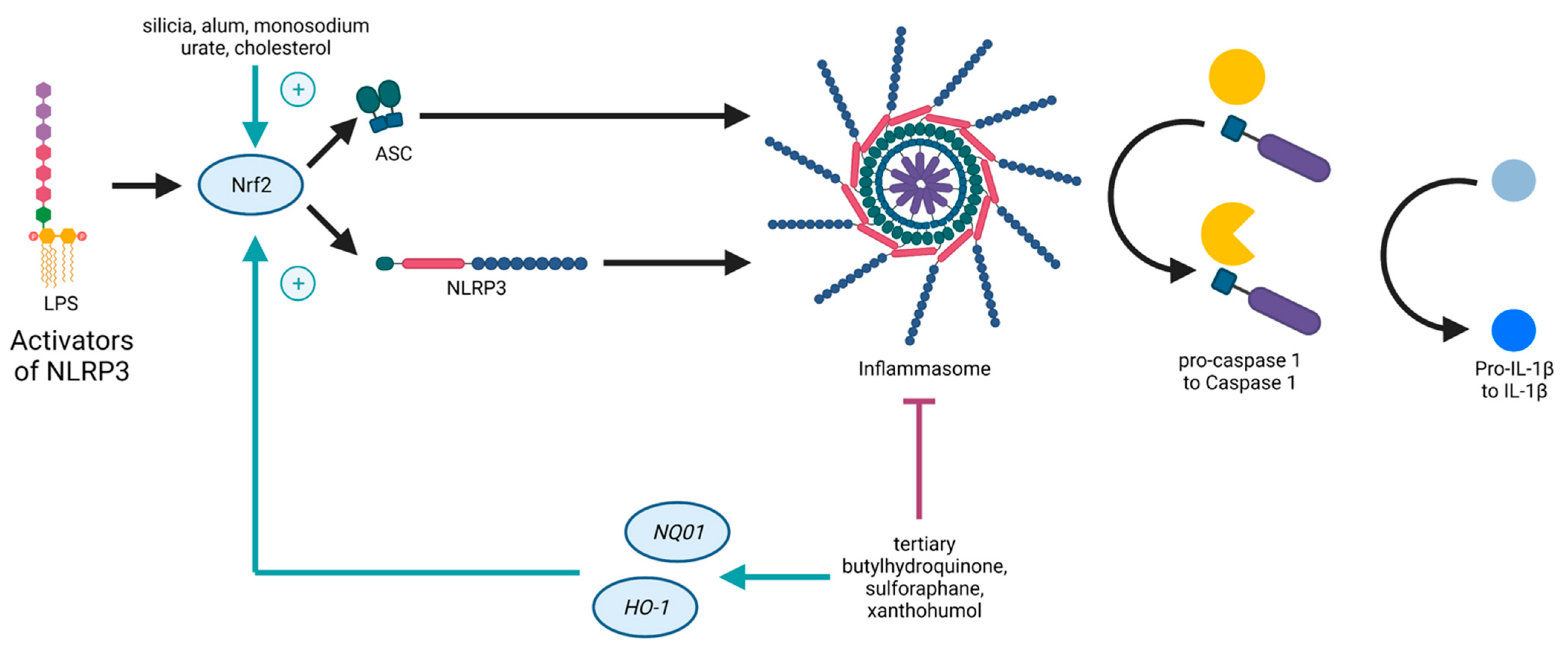
Both the Nrf2- and the NLRP3-pathways are inflammation-associated, stress-induced, and linked to ROS, as well as to NF-κB. On the one hand, ROS is supposed to induce NRLP3 inflammasome activation
[8]. On the other hand, genes expressed due to Nrf2 detoxify ROS
[9]. Nrf2 contributes to NLRP3 inflammasome activation under the condition of oxidative stress
[10]. As mentioned before, NF-κB is necessary for the priming signal of NLRP3 inflammasome activation and also leads to Nrf2 expression
[6]. Furthermore, it was shown that the pathways of Nrf2 and NLRP3 are interconnected in an antagonistic manner
[1], as Nrf2 activation by Nrf2-activating compounds (such as tertiary butylhydroquinone, sulforaphane, and xanthohumol) is accompanied with NLRP3 inflammasome inhibition
[11], providing evidence for novel treatment options against inflammatory disorders. Studies demonstrated that NLRP3 inhibition due to Nrf2 activation is accompanied with a reduction of NF-κB activation
[12][13]. Carbon monoxide, generated in the catalysis of
HO-1, is a negative regulator of NLRP3 inflammasome activation
[14], and thus, inhibits pyroptosis
[15]. However, Nrf2 activated by cholesterol crystals or monosodium urate crystals promotes the activation of the NLRP3 inflammasome
[11] (
Figure 1).
Overall, activation of the host immune response, and further, of inflammation play a crucial role in the development of many chronic diseases. As a pathophysiologic starting point of these processes, several intracellular multimeric protein complexes that activate inflammatory cascade-inducing caspases, the inflammasomes, were identified. There has been recent progress in understanding the role of the NLRP3 inflammasome in oral and systemic diseases. In the field of dentistry, however, evidence regarding the effects of this inflammasome and its potential inhibition, as well as activation due to Nrf2, is missing.
2. The Oral Microbiome
As the initial part of the digestive tract, the oral cavity is the first part of the human microbiome and is followed by the esophagus, stomach, intestine, and colon. More than 700 bacterial, archaeal, viral, and fungal species inhabit the oral cavity and provide the so-called oral microbiome
[16][17][18].
With the achievement of the Human Microbiome Project in 2008, the role of the microbiome in human diseases has become attractive for scientists. Most of the microbiome research focused on the gut microbiome at first, but studies on other organs, i.e., the oral cavity, are increasing progressively. Especially in the field of dentistry, the oral microbiome is a fundamental part of good dental and oral health. With the Human Oral Microbiome Database (HOMD;
www.homd.org; 01/09/2022), extensive information on the approximately 700 predominantly bacterial species of the oral cavity should be provided to the scientific community.
Among digestive organs, the oral cavity is a place of tremendous heterogeneity, due to the presence of teeth and various tissue compositions. Thus, many different niches, including surfaces of the teeth, tongue, cheeks, palate, and tonsils, occupied by specific organisms, form varied environmental compositions with different functional characteristics
[16][19][20]. Dental biomaterials, or prostheses and implants create supplementary locations for biofilm formation
[21][22][23]. In addition, the divergence of the composition of each niche is positively correlated with the periodontal pocket depth and periodontitis progression
[24].
As one of the first bacterial compositions, dental plaque has been described as a complex, polymicrobial, and highly structured biofilm
[25][26]. In the 20th century, researchers discovered species of
Streptococcus mutans [27], which are usually the first pioneering microorganisms of the oral cavity, and the specific periodontal pathogens
Aggregatibacter actinomycetemcomitans (
A. actinomycetemcomitans)
[28],
Porphyromonas gingivalis (
P. gingivalis),
Treponema denticola, and
Tannerella forsythia (
T. forsythia)
[29].
Before birth, the toothless oral cavity of a fetus is sterile and becomes colonized by a common bacterial flora by passing through the birth canal
[30][31]. With the eruption of teeth, a new habitat for microorganisms occurs due to the teeth themselves, and furthermore, through the gingival crevice, which is nourished by the gingival crevicular fluid.
Key to a healthy oral cavity, according recent studies, are microorganisms that can be classified into six phyla, i.e.,
Firmicutes, Bacteroidetes, Proteobacteria, Actinobacteria, Spirochaetes, and
Fusobacteria [32][33][34].
In healthy individuals, a dynamic balance between the microbiome and the host produces important benefits, i.e., controlling the cardiovascular system, defending against potential pathogens, maintaining a healthy digestive tract, developing and supporting host defense functions, and having anti-inflammatory properties
[19][35]. If this homeostasis fails, prolonged dysbiosis and chronic inflammation develop, potentially leading to microbial changes and inflammatory disease complications. To provide oral health of the microenvironment in the biofilm on teeth, the degree of inflammatory response of the tissues in contact with these biofilm is of great importance
[36].
According to recent studies, an imbalance or dysbiosis of the oral microbiome is related to dental caries or periodontitis
[37], oral cancer
[38], inflammatory bowel disease
[39], and systemic diseases, including Alzheimer’s disease
[40], cardiovascular diseases
[41], atherosclerosis
[42], and colorectal, pancreatic, and other cancers
[43][44][45].
A potential microbiome procarcinogenic mechanism derives from TLR-mediated microbial pattern recognition. As TLRs play a crucial role in the interplay between the microbiome and immunity, they stimulate proinflammatory signaling pathways via NF-κB activation
[46], induced by oral microbiota dysbiosis
[42]. As NF-κB leads also to Nrf2 activation
[6], it might be hypothesized that Nrf2 is critical for inhibiting an exaggerated immune response leading to inflammatory diseases.
Furthermore, it has been speculated that Nrf2 has protective effects against inflammatory bowel disease, a chronic and inflammatory disease of the gastrointestinal tract
[47].
Poor oral hygiene facilitates bacterial translocation to other body sites by transmigration from oral draining lymph nodes to other lymphoid organs, i.e., the gut
[48], which means that the oral microbiome and colonic microbiome are physically connected
[49]. Nrf2 is known to be part of gut development
[50] and maintains its regular functionality
[3].
Inflammation, by the specific activation of the NLRP3 inflammasome, is an important contributor to the development of atherosclerotic cardiovascular disease. Baragetti et al.
[51] evaluated this complex pathway to be activated by a number of factors, such as unhealthy nutrition, altered gut, and changed oral microbiome composition.
Moreover, a relevant role of oral microbiome dysbiosis has recently been demonstrated, since it can induce systemic diseases and worsen the metabolic parameters of chronic diseases. Additional studies are expected to confirm that an oral anti-inflammatory treatment and improved oral hygiene can reduce the incidence of diseases. In addition, it is of interest whether the anti-inflammatory effect of Nrf2 may affect the oral microbiome in a protective manner or not.
Overall, innovative development of molecular genetics in DNA sequencing approaches enables us to gather fundamental issues of the oral microbiome and its association with oral and systemic health. Further studies may lead to promising dental interventions to manage the bacterial composition of the oral microbiome, as it has been viewed as an important factor affecting human microbiota.
3. Periodontitis
Periodontitis (PD) is a common chronic inflammatory disease that is caused by bacterial infection in the subgingival microbiome and affects the periodontal tooth-supporting tissues of select teeth or rarely the entire oral structure (gingiva, periodontal ligaments, and alveolar bone). Due to the persistence of periodontal pathogens and an imbalance of the immune response that they encode, PD is characterized by periodontal attachment loss, bone resorption, and can finally lead to tooth loss
[52]. Besides tooth loss, PD can influence systemic health, when oral microorganisms enter the bloodstream by crossing damaged oral mucosa
[42]. Consequently, PD could affect systemic diseases, i.e., cardiovascular disease
[53], rheumatoid arthritis
[54], type 2 diabetes
[55], and cancer
[56]. The main function of the human immune system is to differentiate between commensal bacteria, related to a commensurately oral microbiome, and pathogenic bacteria. Thus, in a healthy balance and established homeostasis, common microorganisms interact with the immune system without provoking a proinflammatory response
[57].
The most likely cause for PD to occur is due to an accumulation of pathogenic bacteria on the tooth surfaces and in the gingiva, followed by inflammation
[58] caused by activation of signaling pathways in PRRs
[59], and thereby, generation of proinflammatory cytokines. In recent years, studies have shed light on the processing steps of these cytokines. Inter alia, this process is dependent upon an intracellular innate immune sensor, the NLRP3 inflammasome that also might influence the PD activity, in response to various bacterial, physical, and chemical agents
[58][60][61].
The IL-1 family of cytokines, such as interleukin-1β (IL-1β) and interleukin-18 (IL-18), are proinflammatory cytokines, which are involved in the pathogenesis of several bone-affecting inflammatory diseases. Furthermore, they mediate bone loss when produced unbalanced. An unbalanced production is due to higher recruitment and differentiation of osteoclasts in the tissues via activation of the receptor activator of nuclear factor kappa-B ligand (RANKL) in osteoblasts
[62][63]. Above all, as an osteoclastogenic factor, RANKL regulates the formation and activity of osteoclasts and upregulates alveolar bone loss
[64]. It has been demonstrated that NLRP3 deficiency can significantly decrease RANKL, indicating the relevance of the NLRP3 inflammasome in supporting osteoclast-genesis in PD
[59]. Studies demonstrated IL-1β to be a potential marker of this disease, due to its higher level in serum, saliva, and gingival tissue of PD patients
[65][66][67]. In the gingival tissue and crevicular fluid of patients with PD, increased expression of IL-1β and IL-18 has been positively correlated with increased expression of
NLRP3 mRNA
[58]. Moreover, an increased expression of
NLRP3 mRNA in the oral epithelium
[68] and in the saliva
[69] of patients has been found with the simultaneous downregulation of NLRP3 inhibitors
[68][70]. Many studies demonstrated that the NLRP3 inflammasome is involved in the development of gingival inflammation and subsequent bone loss, due to an exaggerated immune response
[59][58][60][68][69][71]. In a murine model with bacterial plaque-retentive ligatures placed around the teeth, Marchesan et al.
[72] further support the NLRP3 upregulation in experimental periodontitis.
In response to several bacterial ligands acting as PAMPs, e.g., lipopolysaccharide (LPS)
[73][74], peptidoglycan
[75], bacterial and viral RNA
[76][77], and flagellin
[78], NLRP3 regulates the maturation and secretion of proinflammatory cytokines, like IL-1β
[79] via proCASP1. Furthermore, an upregulation of the NLRP3 inflammasome complex leads to an increase in the genesis of IL-1β
[58][70] and IL-18
[60][80][81].
IL-1β is mainly produced in monocytes, which are declared to express
NLRP3 mRNA
[82][83]. Sutterwala et al.
[73] found that this process was highly induced by bacterial LPS. On the other hand, there was no production of IL-1β in NLRP3-deficient macrophages, even though bacterial stimulations occurred
[75][84][77][85].
Treatment with an IL-1 receptor antagonist of patients with rheumatoid arthritis cancelled clinical symptoms, suggesting a cause–effect relationship between IL-1β production and the development of the disease
[86]. Based on an inhibition of the NLRP3 inflammasome, some studies also presented therapeutic pathways in the treatment of experimental PD
[87][88].
Moreover, Nrf2 has been demonstrated to directly suppress transcription of NLRP3-associated genes, including
pro-IL-1β, and
pro-IL-1α [89][90], suggesting Nrf2 to be a potential therapeutic inhibitor of PD.
Delineating the likely role of several oral microbiota associated with the development of PD is rather complex. Among the thousands of bacterial species in the oral cavity, few Gram-negative anaerobic bacteria were related to PD genesis. Periodontopathogen species dispose of several virulence factors that allow them to survive in the host environment by selectively adapting the host’s immune-inflammatory response. The “red cluster” of periodontopathogenic bacteria, consisting of
P. gingivalis,
Treponema denticola,
Prevotella intermedia,
A. actinomycetemcomitans, and
T. forsythia, contribute to the initiation and progression of severe PD
[91][92].
Teles et al.
[93] showed a positive correlation between these bacterial species and the overexpression of proinflammatory cytokines, i.e., IL-1β and IL-18. Consequently, as PAMPS, these bacteria are attributed to have an impact on the etiology and progression of PD by activating inflammasome activity and controlling the NLRP3-mediated inflammatory response in PD
[94]. Furthermore, an in vitro study has demonstrated periodontopathogenic bacteria, such
P. gingivalis,
A. actinomycetemcomitans, or
Fusobacterium nucleatum (F. nucleatum), to be responsible for an increased expression of NLRP3
[95]. Several signaling pathways have been demonstrated to generate and promote the occurrence of PD. In this context, it is essential to know and understand them, as modulating them may be the key in preventing or treating PD.
4. Periapical Periodontitis
Besides PD in the traditional sense of term, i.e., gingival PD, periapical PD is one of the most common inflammatory diseases in adults. In response to caries, tooth fracture, or trauma, oral microorganisms can enter the initial sterile tooth pulp and trigger inflammation, which may result in pulp necrosis [174,175]. Symptoms are varied, implicating sensitivity to pressure or cold, pain, periapical radiolucency, and edema [176]. There is a distinction between acute and chronic periapical PD showing different symptoms [175]. Most of endodontic bacteria are located in the root canal [177]; thus, the therapy of choice is a root canal treatment, aiming to remove the inflamed dental pulp [178,179]. Surgical apicoectomy is required when endodontics is insufficient and the inflamed part of the bone includes the tooth apex [180]. Etiology of this odontogenic infection is due to bacterial species and their virulence, as well as the interaction with immunological host responses [175]. It was shown that apical PD is responsible for generating cytokines by recruiting inflammatory cells, i.e., host immune response to inflammatory processes [181].
The most common pathogen in periapical PD was demonstrated to be Enterococcus faecalis (E. faecalis), a Gram-positive coccus [182,183,184]. It was already shown that E. faecalis is able to promote CASP1 activation and pro-IL-1β expression, which subsequently increases IL-1β levels [185]. Moreover, increasing IL-1β production during periapical PD [186] might be associated with an interplay between this inflammatory disease and the NLRP3 inflammasome.
5. Oral Squamous Cell Carcinoma
With an incidence of 90%, oral squamous cell carcinoma (OSCC) is the most common oral cancer
[96] with a low 5-year survival rate of only 30%
[97]. Despite the frequent inspection of the oral cavity by dentists assuming patients’ responsibility for oral health, most OSCC lesions were missed at an early stage, which may explain the high mortality rate. OSCC is known to occur three times more frequently in men than in women
[98]. Besides smoking and alcohol consumption
[99], risk factors of OSCC also include chronic inflammation
[100]. Several studies have shed light on inflammation as a possible cancer basis, as Hussain et al.
[101] already determined in 2003 that inflammation is the cause of one in four cancers. Furthermore, oral bacterial species are reported to be responsible for these inflammatory disorders via influencing key processes, which may be contributing to oral carcinogenesis
[102][103].
As already described before,
P. gingivalis and
F. nucleatum induce the development of proinflammatory cytokines, at least partially, through the NLRP3 pathway. Therefore, it was proved that these periodontopathogenic bacteria are potential etiological agents for oral cancer
[104]. Yang et al.
[105] provided evidence that
Fusobacteria can be associated with cancer staging of OSCC. Interestingly, Tezal et al.
[106] evaluated patients whom had never used tobacco and alcohol, but suffered from PD. These patients showed a higher probability of 32.8% for poorly differentiated OSCC than patients of good oral health at 11.5%. A very recent study by Yao et al.
[107] from 2021 developed a new mechanism connecting periodontopathogenic bacteria (
P. gingivalis and
F. nucleatum) and OSCC, by showing that these bacterial species upregulate NLRP3 expression and drive inflammasome activation. It was examined that this pathway can level up tumor growth and tumor proliferation.
P. gingivalis has been reported to be carcinogenic. Studies demonstrated a positive correlation between
P. gingivalis infection and size or invasiveness
[108][109], as well as a late tumor–node metastasis (TNM) stage, and low differentiation
[110] of head and neck squamous cell cancer (HNSCC). In addition, Sinha et al.
[111] first demonstrated that
P. gingivalis is enriched in feces from patients with colorectal cancer. Wang et al.
[112] later confirmed that
P. gingivalis can be held responsible for enhancing colorectal cancer, due to the activation of the NLRP3 inflammasome.
Taken together, oral health and, subsequently, the oral microbiome and its interplay with the host immune response may play a critical role in the development of OSCC. The importance of an efficient and healthy oral microbiome has been underlined by developing a novel strategy of detecting OSCC due to the utilization of saliva, which makes the oral microbiome a noninvasive diagnostic tool
[113][105].
Standard therapy of OSCC is a treatment using 5-Fluorouracil (5-FU), which inhibits pyrimidine metabolism and DNA synthesis. It was determined that treatment with 5-FU leads to increased intracellular ROS and is ascertained to upregulate NLRP3 and IL-1β expression in human OSCC cell lines. This subsequently mediates a chemoresistance of OSCC to 5-FU lying on multiple aspects, suggesting tumor-associated macrophages to be responsible. Moreover, survival rates of patients decreased with higher NLRP3 expression, and NLRP3 deficiency enhanced the antitumor effect of 5-FU
[114]. This might confirm that NLRP3 is responsible for not only the progression of OSCC but also its limited treatment effectiveness.
As NLRP3 is likely to be critically involved in OSCC occurrence, progression, and proliferation, few studies have shed light on possible strategies for oral cancer treatment, regarding the NLRP3-inflammasome. MicroRNAs are known to act as regulators of carcinogenesis in general. Feng et al.
[115] implicated microRNA-22 to inhibit OSCC proliferation due to interference of the NLRP3 pathway.
So-called dietary exosome-like nanoparticles extracted from mushrooms
[116] or ginger rhizomes
[117] were identified as inhibitors of the NLRP3 inflammasome.
Yang et al.
[118] showed that also bitter melon-derived extracellular vesicles (BMEVs) may downregulate the NLRP3 activation and, moreover, reduce the drug resistance of 5-FU. Additionally, BMEVs could inhibit OSCC proliferation due to the development of reactive oxygen species.
BAY 11-70082 is a sulfonic derivative and an inhibitor of NF-κB, known for having anticancer and anti-inflammatory effects
[119]. Scuderi et al.
[120] found that BAY 11-70082 could downregulate NLRP3 activation, and further, reduce tumor mass in mice.
In summary, OSCC treatment targeting NLRP3 is highly promising, and thus, necessitates further research.
It is known that oxidative stress plays an important role in the development of cancer. Nrf2, as an oxidative stress marker, was found to be associated with carcinogenesis and progression of OSCC
[121] when hyperactive, while it inhibited carcinogenesis of normal cells
[122]. This might suggest a prognostic value of Nrf2 and its potential role as therapeutic target.