Cationic liposomes (CLs) are assemblies of lipids with an overall cationic (positive) charge that is contributed by the sum of the charges of the lipid headgroups. In particular, they are spherical bilayer assemblies of cationic lipids or a mixture of neutral and cationic lipids.
CLs effective carriers of a variety of therapeutics. Their applications as vectors of nucleic acids (NAs), from long DNA and mRNA to short interfering RNA (siRNA), have been pursued for decades to realize the promise of gene therapy, with approvals of the siRNA therapeutic patisiran and two mRNA vaccines against COVID-19 as recent milestones. The long-term goal of developing optimized CL-based NA carriers for a broad range of medical applications requires a comprehensive understanding of the structure of these vectors and their interactions with cell membranes and components that lead to the release and activity of the NAs within the cell.
- cationic liposomes
- nucleic acids
- nanoparticles
- hydrophobic drug delivery
- gene therapy
- small-angle X-ray scattering
- homing peptide
- affinity targeting
1. Introduction
Amphiphilic molecules, and so i.e.on, molecules with a polar, hydrophilic headgroup and a hydrophobic tail or tails, spontaneously self-assemble in water, primarily due to hydrophobic interactions [1]. The resulting assemblies expose the headgroups to the aqueous environment while avoiding exposure of the tails, which instead form their own hydrophobic environment. This understanding, which is now commonplace, originated only after Bangham and Horne discovered liposomes (also referred to as unilamellar or multilamellar vesicles; Figure 1 ) while studying suspensions of phospholipids by electron microscopy [2]. Liposomes, consisting of closed assemblies of bilayers of lipids, resembled cell membranes in the electron micrographs, thereby confirming that the makeup of these biological structures is lipid-based. Bangham and Horne’s work further demonstrated that lipid bilayer membranes can accommodate hydrophobic molecules while forming a barrier for polar, hydrophilic molecules. Thus, these studies also confirmed the hypothesis that the lipids of plasma membranes provide the permeability barrier that is essential to biological membrane function.
In the wake of the landmark paper by Felgner et al. [3] which introduced cationic lipids as functional carriers of DNA, countless cationic lipids have been synthesized and studied [4][5][6][7][8][9][10][11][12][13][14][15][16]. In addition, numerous aspects of the formation and action of complexes of CLs with DNA and other nucleic acids (NAs) have been investigated [17][18][19][20][21][22][23][24][25][26][27][28]. These worldwide research efforts with CL vectors have the aim of improving efficacy both in vitro (at the cell level) and in vivo.
The importance of charge in preparing effective CL-based NA vectors was appreciated early on. In addition to the fact that complex formation with CLs protects NAs from degradation, an early rationale for utilizing cationic rather than neutral or negative liposomes to deliver DNA was that CL–DNA complexes designed to have an overall positive charge (i.e., with a lipid/DNA charge ratio > 1) would electrostatically adsorb to anionic mammalian cells, thus leading to cellular binding and more efficient uptake [29]. Here, the lipid/DNA charge ratio refers to the number of charges on the lipid divided by the number of charges on the DNA (under the conditions that the lipid and DNA are combined).
Establishing other structure–activity relationships for cationic lipids for NA delivery is more challenging than for small-molecule drugs. Instead of a single molecule interacting with, e.g., the binding pocket of a protein, a large number of lipids joins together in a dynamic assembly before interacting with the NA to form the active principle that engages with the target tissue or cell. In addition, the local environment may affect the CL–NA assembly and the interactions with its target. Thus, changes to the structure of the lipid may affect the efficacy of the NA vector in ways that are amplified by the process of self-assembly. This enItry aim was to provide a guide for parsing through the overwhelming number of lipids and formulations by relating the lipid structure to properties of lipid self-assembly. Some of these properties are only peripherally known to chemists designing these lipids because they stem from the biophysics and physical chemistry of lipids as well as from colloid science.
2. Lipid shape and membrane curvature elastic energy determine the structures of lipid self-assemblies
It has been appreciated in surfactant and lipid science for over four decades that the shape of an amphiphile plays a major role in determining the structure of its self-assemblies [1][30][31][32][33]. Figure 2 illustrates the concept of lipid shape and the resulting assemblies with molecular models of a few lipids for illustration. The area of the lipid’s headgroup relative to that of the tails determines the lipid’s shape, and arrangement of these shapes with the constraint of exposing tails only to themselves and headgroups to the exterior aqueous environment naturally leads to the self-assembled structure. These assemblies, in turn, constitute the lipid building blocks of resulting CL–NA complexes.
A quantitative (but nonetheless semiempiric) parameter for describing lipid shape and the resulting assemblies is the “packing parameter”, v/a0lc. This dimensionless parameter is the ratio of the average area of the tails, expressed as volume of the tails (v) over their maximum effective length (lc) and the optimal headgroup area (a0) [1]. For the lipids in Figure 2, the packing parameter increases from left to right, with v/a0lc=1 for lipids with cylindrical shape.

The physical formalism to describe lipid membranes and their elastic properties uses the membrane curvatures, defined as the inverse of the membrane radii (Ci=1/Ri) in two orthogonal directions (Figure 2). For example, C1=C2=0 for a flat membrane; C1=C2=1/Rsph for a spherical membrane with radius Rsph; and C1=0, C2=Rcyl for a cylindrical membrane with radius Rcyl. Negative curvature is also possible, e.g. for the surface of an inverse micelle (Figure 2, top left). The shape of a lipid determines the spontaneous (in other words, preferred) curvature of the membrane, termed C0. Introducing further the membrane modulus κ (a measure of the bending stiffness of the membrane) and the Gaussian modulus κG (a measure of the propensity for (κG>0) or resistance to (κG<0) forming saddle shapes), the elastic free energy of a membrane per unit area, E/A, is
|
E/A = 0.5 κ (C–C0)2 + κG C1C2, |
(1) |
with C=C1+C2 being the mean curvature of the membrane and C1C2 termed the Gaussian curvature of the membrane [34][35][36]. The elastic moduli κ and κG can be related to the interactions between neighboring lipids in the membrane using harmonic spring models [37][38].
The first term in Eq. 1 describes the free energy cost of bending a membrane away from its spontaneous curvature, which increases as the membrane stiffness (κ) increases. As mentioned above, the shape of a lipid determines C0 [1][39]. Intuitively it may be easier to consider the spontaneous radius of curvature, R0, in some cases. Lipids with a headgroup area that is approximately equal to the projected area of their tails have a cylindrical shape. This corresponds to a spontaneous curvature C0=0 for the bilayer (Figure 2, center), and these lipids tend to assemble into lamellar bilayer structures such as lamellar lyotropic phases (at low water content) or large uni- and multilamellar vesicles (in excess water). Examples of such lipids are the neutral lipid DOPC (as well as other related phosphocholines with saturated tails) and the monovalent cationic lipid DOTAP. The molecular models in Figure 2 show that the steric size of the headgroup of DOTAP is much smaller than that of DOPC (or DOPE, see below), but the charge of the headgroup and the electrostatic repulsion between lipids that it induces as well as its hydration must be taken into account when considering the shape of the lipid in an aqueous environment.
The shape of a lipid with a headgroup area that is smaller than the tail area is described as an inverse cone. For lipids of this shape, C0<0, and they tend to form inverse micelles (Figure 2, left) that assemble further into structures such as the inverse hexagonal (HII) phase [39][40] for inverse cylindrical micelles or cubic phases for inverse spherical micelles. A well-known example of such lipids is DOPE, which differs from DOPC only in bearing an amino- instead of a trimethylammonium-group in its headgroup. This results It is in a larger hydration of the DOPC versus the DOPE headgroup and thus a difference in lipid shape, again highlighting that the structure of lipids must be considered in the context of their surroundings.
Instead of a decrease in headgroup size, an increase in the average cross-sectional area of the lipid tails can also change lipid shape from cylinder to inverse cone. An example of this is the lipid DLinPC, which bears two cis double bonds in its linoleoyl tails, compared to the single double bond in each tail of DOPC. As illustrated in Figure 2, the additional bend in the tail cannot be compensated for by gauche conformations in the rest of the chain and the tail area increases. Consistent with this, CLs containing DLinPC showed a propensity to form the inverse hexagonal phase [41]. Similarly, increasing chain unsaturation facilitated formation of the inverse hexagonal phase for a series of cationic lipids with C18 tails [42]. Of these lipids, the one with linoleyl tails (DLin-DMA), has a structure very similar to the cationic lipid used in the patisiran formulation (DLin-MC3-DMA; note the tails with two cis double bonds). It is intriguing to note in this context that the branching of the tails of the cationic lipids used in the mRNA vaccines against COVID-19 also suggest that they have an increased cross-sectional area.
Cone-shaped lipids have a headgroup with an area that is larger than that of the tail. In this case, C0>0, and the lipids usually assemble into cylindrical or spheroidal micellar structures (Figure 2, right). This shape is common for amphiphiles with a single tail, but their high toxicity prevents their use in therapeutics. Lipids with two tails can be cone-shaped if they have a headgroup with high charge and/or large steric size. An extreme example is the custom-synthesized lipid MVLBG2 65,66, with a headgroup that bears 16 positive charges at full protonation. Lipids with a more moderate charge, such as DOGS [43] and MVL5 [44][45], have also shown a propensity for micellar structures. The headgroup of PEG-lipids is large because of the steric size of the polymer chain (depending on PEG length), and lipid mixtures containing them also form micellar structures, depending on the fraction of PEG-lipid [46][47][48].
As mentioned above, the environment of a lipid must be taken into account when considering its shape, and changing conditions can lead to a change in shape and thus the structure of the lipid assembly. One example of this are changes in pH, which may increase or reduce the charge of a lipid’s headgroup, depending on the lipid structure. Another is the salt concentration in the aqueous environment; higher salt concentrations screen electrostatic interactions, effectively reducing the headgroup size.
The spontaneous curvature of a mixture of lipids is equal to the sum of the spontaneous curvatures of the individual lipids, C0,i, weighted with their molar fraction, xi, provided that complete mixing of lipids in the membrane takes place. Thus, for a mixture of two lipids, C0= x1C0,1 + x2C0,2, with xi=ni/(n1 + n2) and n1, n2 the number of the two lipids in the membrane. An illustration of this is provided by mixtures of cone-shaped MVLBG2 with cylindrical DOPC. These mixtures form sheets (vesicles), cylindrical micelles that shorten with increasing content of MVLBG2, and spherical micelles at low, intermediate, and very high contents of MVLBG2, respectively [49]. An example on the other side of the curvature spectrum are mixtures of DOPE and DOPC, which form lamellar or inverse micellar structures depending on their composition [33].
The second term in Eq. 1 contains the Gaussian modulus, κG, and the Gaussian curvature, C1C2. Depending on the sign of their Gaussian modulus, membranes will prefer to form shapes of either positive or negative Gaussian curvature to minimize their elastic free energy. Examples of shapes with a positive Gaussian curvature (C1C2>0) are the outer (C1>0, C2>0) and inner (C1<0, C2<0) monolayer of a spherical vesicle, while flat bilayers (C1=C2=0) and cylindrical micelles (C1>0, C2=0) have a Gaussian curvature of zero. Membranes with a positive Gaussian modulus κG>0 will favor saddle-shaped surfaces with C1C2<0. These include the surfaces of bicontinuous cubic phases (Figure 6, left) and membrane pores (Figure 6, right; note the resemblance of the shape to a saddle) [35][39][50]. Thus, a consideration of membrane elasticity suggests that cubic phase-forming lipids (i.e., with κG>0) favor formation of pores (resulting from fusion of two membranes that face each other in close proximity) [50][51][52]. Because escape from the endosome after being internalized by the cell, which requires pore formation, is a major barrier to successful NA delivery, this is a highly relevant insight to the development of efficient CL-based NA vectors.
3. The Self-Assembled Structures of CL–NA Complexes
While their opposite charge makes it appear intuitive that cationic liposomes and anionic NAs form complexes, the fact is that both already are associated with neutralizing counterions in solution. The primary driving force for their spontaneous self-assembly is the entropy gained by the release of positive counterions that are tightly bound to DNA (referred to as Manning condensation [53]) and negative counterions near the cationic liposome surface within the Guoy–Chapman layer [3]. As the cationic membranes neutralize the phosphate groups on the DNA, those small counterions gain in entropy because they are now free to diffuse and no longer bound to the DNA or cationic membranes. This driving force is also important in the assembly of other oppositely charged macro-ions, e.g. cationic polymers and DNA [54] or CLs and anionic proteins.
Initially, complexes of CLs and DNA were thought to consist of intact liposomes associated with DNA in a “bead-on-string” or “spaghetti and meatballs” structure [55]. Many subsequent studies have shown that such highly disordered assemblies are at best a short-lived intermediate in the process of CL–DNA complex formation. Ultimately, a complete topological transition from liposomes into collapsed condensates in the form of distinct liquid crystalline self-assemblies occurs [56][57][58][59][60]. The internal structure of the CL–DNA complexes as initially revealed by synchrotron small-angle x-ray scattering (SAXS) studies [56] is consistent with images from cryogenic electron microscopy studies [60][61][62].
As mentioned above, membrane shape is often determined by the spontaneous curvature of the membrane, which is related to the molecular shape of the constituent lipids. The shape of the membrane component, in turn, often determines the structure of the resulting CL–NA complex. Bilayer sheets, reflecting a spontaneous curvature of C0=0, as building blocks give rise to the most common phase of CL–DNA complexes. This phase was also the first one to be discovered [60][56]: the lamellar, LαC, phase (Figure 3, Top), consisting of a multilamellar arrangement of DNA monolayers intercalated (sandwiched) between cationic membranes.
The LαC structure has been observed in CL–DNA complexes containing a broad variety of DNA and CLs formed from a wide variety of cationic lipids. It has also been found in complexes of CLs with siRNA [63] and single-stranded mRNA [64]. Other phases that are closely related to the LαC phase but less relevant to delivery applications have also been reported. Three-dimensional columnar phases are characterized by order of the DNA chains not only within a single layer, but also between layers. Such phases have been observed for complexes of CL membranes in the “gel” phase with chain-ordered lipids with long DNA [65][66][67].
The inverse micellar building blocks resulting from lipids with preferred curvature C0<0, such as DOPE, favor formation of the inverted hexagonal phase of CL–DNA complexes (Figure 3, Middle). In this phase, termed HIIC, DNA chains occupy the aqueous interior of inverse cylindrical micelles, and those micelles are arranged on a hexagonal lattice, forming a 2D columnar liquid crystalline phase.
Complexes of DOTAP/DOPE CLs with long linear DNA [68], plasmid DNA [69] as well as with siRNA [63] form inverse hexagonal phases at sufficiently high membrane content of DOPE. This supports the hypothesis that the membrane structure, guided by the lipid shape, is the main determinant of the structure of CL–NA assemblies.


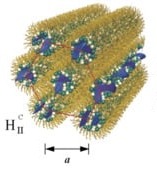
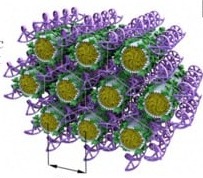
When the lipid assemblies are cylindrical micelles (for lipids with positive preferred curvature, C0>0), CL–DNA complexes may form the hexagonal (HIC) phase (Figure 3, Bottom). In this liquid crystalline structure, the cylindrical lipid micelles form a 2D hexagonal lattice. They are surrounded by the DNA, which forms a three-dimensionally continuous substructure with honeycomb symmetry [58]. This is an interesting contrast to the isolated DNA rods (1D) and sheets (2D) in the HIIC and LaC phases, respectively (Figure 3, Top and Middle).
The HIC phase was first observed in a narrow composition range of CL–DNA complexes containing ≈25 mol% MVLBG2 (with the remainder DOPC) in their membranes. MVLBG2 is a highly charged (16+) multivalent cationic lipid with a dendritic headgroup [70][15]. A distorted HIC phase was observed at higher contents of MVLBG2 and other highly charged (8+) dendritic lipids [49]. However, the headgroups of most other, even multivalent (up to 5+) cationic lipids, appear to be too small to force the formation of the cylindrical micelles that make up these phases. For MVLBG2/DOPC–DNA complexes with higher contents (30–50 mol%) of MVLBG2, SAXS reveals a distorted hexagonal HIC phase [49].
In some cases, the incorporation of NA disrupts the preferred phase of the lipid component because it can not otherwise be accommodated in the assembly. For example, the lipid GMO [71] readily forms bicontinuous cubic phases such as the gyroid QIIG with space group Ia3d; this holds true even when GMO is mixed with (relatively small amounts of) cationic lipids such as DOTAP or MVL5 [59][72][73]. Both of these cubic phases with added cationic lipid are able to incorporate functional siRNA to generate a novel bicontinuous double gyroid cubic lipid phase, with the siRNA residing its two water channels (Figure 4) [59][72]. The name of the phase indicates the fact that both the water and the lipid subphases are continuous in three dimensions, and that there are two independent water channels of gyroid symmetry (Ia3d space group). A gyroid cubic structure has also been proposed for siRNA complexes of a mixture of GMO with divalent cationic gemini lipids [74]. More recently, cubic CL–siRNA complexes with Im3m symmetry have been discovered when a small amount of PEG-GMO lipid was added to DOTAP/GMO mixtures at very high GMO content (95 mol%) [75][76].
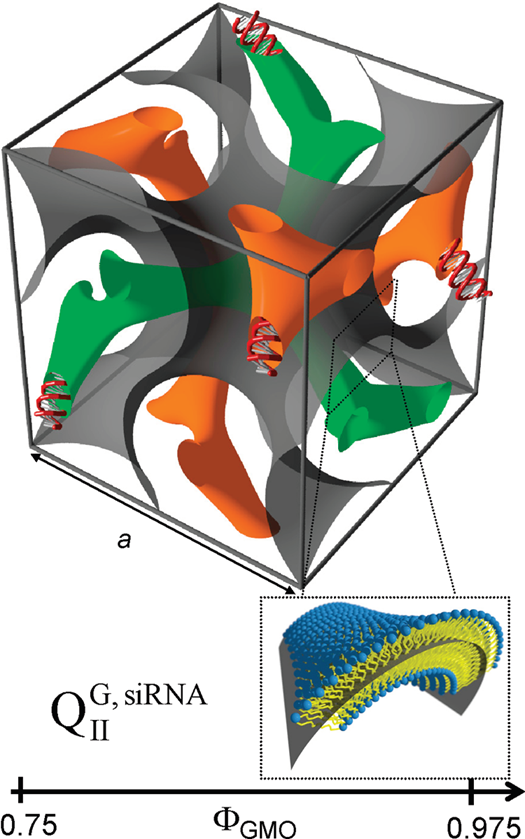
Figure 4. Schematic depiction of the double-gyroid cubic phase of CL–siRNA complexes labeled (QIIG, siRNA). The two intertwined but independent water channels are shown in green and orange. For clarity, the lipid membrane separating the two water channels is represented by a gray surface corresponding to its center (see inset). Note the negative Gaussian curvature of the bilayer, C1C2<0. Reprinted with permission from [59]. Copyright 2010 American Chemical Society.
When CLs of the compositions that form double-gyroid cubic CL–siRNA complexes are mixed with DNA, complexes in the HIIC phase are formed. This likely occurs because the energetic cost of bending the DNA to conform to the highly curved channels of the double-gyroid phase is greater than the cost for rearranging the lipid phase into the related HIIC phase. In fact, studies with a series of short DNA duplexes of varied length and end structure (“sticky”, repulsive, and no overhangs) revealed that stacking of these short pieces of DNA can induce the HIIC phase. Thus, counteracting the stacking with repulsive (nonpairing) overhangs or increased temperature shifted the equilibrium towards the gyroid cubic phase [77].
For a discussion of relations between the the structure of CL–DNA complexes of and their transfection efficiency, the reader is referred to review from which this entry has been adapted (see the bottom of this entry).
4. Cationic Liposomes for The Delivery of Hydrophobic Drugs
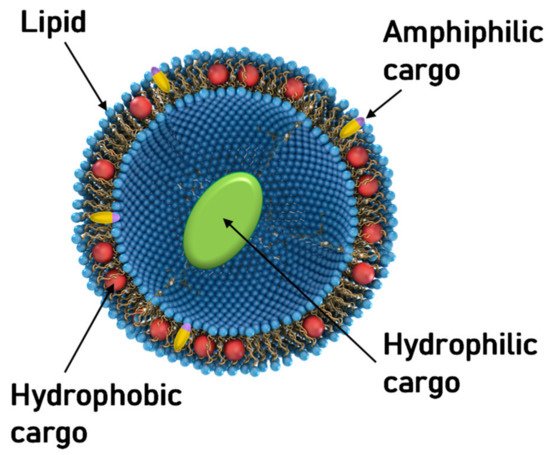
Aside from NA delivery, cationic liposomes have received intense interest as carriers for other classes of therapeutics, in particular hydrophobic cancer drugs ( Figure 1 ) [78][79][80][81][82][83][84]. Many of the principles underlying structure–activity relationships of CL–NA assemblies (such as the effect of lipid shape on assembly structure) that have been outlined in the previous sections are also relevant in this context, even as some intriguing differences exist (e.g., with respect to the effect of PEGylation, see below).
A prominent example of a hydrophobic cancer drug is paclitaxel (PTX), which is among the most widely used chemotherapy drugs to treat ovarian, breast, lung, pancreatic, and other cancers [85][86][87][88][89][90][91][92][93]. Because of their hydrophobicity, these drugs reside within the lipid bilayer rather than in the aqueous interior of the liposome ( Figure 1 , red spheres). This means that they will remain associated with the lipid membranes upon formation of CL–NA complexes, resulting in the exciting potential to combine them with NAs into therapeutics with dual action [94].
Because of their low water solubility, hydrophobic drugs such as PTX are not effective unless formulated with a carrier. Currently used formulations of PTX (such as Taxol® and Abraxane® ); frequently cause hypersensitivity reactions and/or deliver PTX non-discriminately throughout the body [95][96][97]. Therefore, the development of liposomal formulations of PTX with high efficacy is an extremely active field of research [98][99][100][101][102], including in clinical trials [84][102]. Cationic liposomes have been shown to target tumor neovasculature [79][80][81][103][104] and are readily internalized by cells. This makes cationic liposomes desirable vectors for hydrophobic drugs because molecules such as PTX have to reach the inside of cells to unfold their activity.
A common feature of liposomal formulations of PTX is their relatively low loading capacity, at 3 mol% of the lipid content. Improving the PTX loading capacity of liposomal carriers requires enhancing PTX solubilization within the membrane [105]. Recent work showed that there is great potential for achieving this by modifying the lipid tails [41]. PTX membrane solubility in cationic liposomes with tails containing two cis double bonds (linoleoyl tails) was significantly increased compared to tails with one (oleoyl tails) double bond: 8 mol% PTX in the former remained soluble for approximately as long as 3 mol% PTX (the above-mentioned membrane solubility limit) in the latter. Comparison with DOPE-containing formulations suggests that the increase in solubility is likely not caused by the structural change from bilayers to inverse cylindrical micelles ( Figure 2 ) but rather by the enhanced molecular affinity between lipid tails and PTX. Importantly, the efficacy of the PTX-loaded CLs was unaffected by changing the lipid tails in one cell line; in another cell line, the efficacy was even increased two-fold. These findings demonstrate the potential of chemical modifications of the lipid tails: liposomal PTX carriers with increased PTX solubility, at maintained or even increased efficacy, reduce side effects and costs because they require less lipid to deliver a given amount of PTX.
5. Concluding Remarks
As the range of applications of cationic liposomes in the delivery of therapeutics further matures and grows, it becomes ever more important to consider principles from soft matter and biophysics and lipid and colloid science when trying to understand the structure–activities of the cationic and neutral lipids, because the lipids exert their function as an assembly of molecules. The results of ongoing studies investigating the barriers to targeted delivery and the mechanisms of intracellular uptake, transport, and release of these vectors and their interactions with cell components will continue to inform the design and synthesis of optimal lipid carriers of NAs.
Even as the medical applications are just beginning, cationic liposome-based vectors of nucleic acids for gene delivery, gene silencing, and gene editing have found myriad applications in fundamental and applied biological research. Further advances in the delivery systems will only add to these applications, be it in studies of chromosome structure and function through the development of efficient vectors for very large DNA constructs, or in molecular biology studies through improvements in the ability to efficiently deliver NAs into hard-to-transfect cell lines such as T-cells, macrophages and dendritic cells. Other important frontiers of the field are selectively targeting tissues beyond the liver (patisiran) and local delivery (mRNA vaccines). Finally, for cancer therapeutics, the ability of CLs to simultaneously deliver both NAs and hydrophobic drugs in combination therapies holds great promise.
References
- Israelachvili, Jacob N. . Intermolecular and Surface Forces, 3rd ed.; Elsevier: Amsterdam, 2011; pp. 1.
- Bangham, A.D.; Horne, R.W. Negative staining of phospholipids and their structural modification by surface-active agents as observed in the electron microscope. J. Mol. Biol. 1964, 8, 660–668.
- P. L. Felgner; T. R. Gadek; M. Holm; R. Roman; H. W. Chan; M. Wenz; J. P. Northrop; G. M. Ringold; Mark Danielsen; Lipofection: a highly efficient, lipid-mediated DNA-transfection procedure.. Proceedings of the National Academy of Sciences 1987, 84, 7413-7417, 10.1073/pnas.84.21.7413.
- Ponti, F.; Campolungo, M.; Melchiori, C.; Bono, N.; Candiani, G. Cationic lipids for gene delivery: Many players, one goal. Chem. Phys. Lipids 2021, 235, 105032.
- Semple, S.C.; Akinc, A.; Chen, J.; Sandhu, A.P.; Mui, B.L.; Cho, C.K.; Sah, D.W.Y.; Stebbing, D.; Crosley, E.J.; Yaworski, E.; et al. Rational design of cationic lipids for siRNA delivery. Nat. Biotechnol. 2010, 28, 172–176.
- Koynova, R.; Tenchov, B.; Wang, L.; MacDonald, R.C. Hydrophobic Moiety of Cationic Lipids Strongly Modulates Their Transfection Activity. Mol. Pharm. 2009, 6, 951–958.
- Tranchant, I.; Thompson, B.; Nicolazzi, C.; Mignet, N.; Scherman, D. Physicochemical optimisation of plasmid delivery by cationic lipids. J. Gene Med. 2004, 6, S24–S35.
- Labas, R.; Beilvert, F.; Barteau, B.; David, S.; Chèvre, R.; Pitard, B. Nature as a source of inspiration for cationic lipid synthesis. Genetica 2010, 138, 153–168.
- Zhi, D.; Zhang, S.; Cui, S.; Zhao, Y.; Wang, Y.; Zhao, D. The Headgroup Evolution of Cationic Lipids for Gene Delivery. Bioconjug. Chem. 2013, 24, 487–519.
- Wölk, C.; Janich, C.; Bakowsky, U.; Langner, A.; Brezesinski, G. Malonic acid based cationic lipids—The way to highly efficient DNA-carriers. Adv. Colloid Interface Sci. 2017, 248, 20–34.
- Miller, A.D. Cationic Liposomes for Gene Therapy. Angew. Chem. Int. Ed. Engl. 1998, 37, 1768–1785.
- Carrière, M.; Tranchant, I.; Niore, P.-A.; Byk, G.; Mignet, N.; Escriou, V.; Scherman, D.; Herscovici, J. Optimization of Cationic Lipid Mediated Gene Transfer: Structure-Function, Physico-Chemical, and Cellular Studies. J. Liposome Res. 2002, 12, 95–106.
- Bhattacharya, S.; Bajaj, A. Advances in gene delivery through molecular design of cationic lipids. Chem. Commun. 2009, 31, 4632–4656.
- Ewert, K.; Slack, N.L.; Ahmad, A.; Evans, H.M.; Lin, A.J.; Samuel, C.E.; Safinya, C.R. Cationic lipid-DNA complexes for gene therapy: Understanding the relationship between complex structure and gene delivery pathways at the molecular level. Curr. Med. Chem. 2004, 11, 133–149.
- Ewert, K.K.; Evans, H.M.; Bouxsein, N.F.; Safinya, C.R. Dendritic cationic lipids with highly charged headgroups for efficient gene delivery. Bioconjug. Chem. 2006, 17, 877–888.
- Maier, M.A.; Jayaraman, M.; Matsuda, S.; Liu, J.; Barros, S.; Querbes, W.; Tam, Y.K.; Ansell, S.M.; Kumar, V.; Qin, J.; et al. Biodegradable Lipids Enabling Rapidly Eliminated Lipid Nanoparticles for Systemic Delivery of RNAi Therapeutics. Mol. Ther. 2013, 21, 1570–1578.
- Chesnoy, S.; Huang, L. Structure and function of lipid-DNA complexes for gene delivery. Annu. Rev. Biophys. Biomol. Struct. 2000, 29, 27–47.
- Ewert, K.; Evans, H.M.; Ahmad, A.; Slack, N.L.; Lin, A.J.; Martin-Herranz, A.; Safinya, C.R. Lipoplex Structures and Their Distinct Cellular Pathways. In Advances in Genetics, Vol. 53: Non-Viral Vectors for Gene Therapy, 2nd ed.; Huang, L., Hung, M.C., Wagner, E., Eds.; Elsevier; Academic Press: San Diego, CA, USA, 2005; pp. 119–155.
- Ewert, K.; Ahmad, A.; Evans, H.; Safinya, C. Cationic lipid-DNA complexes for non-viral gene therapy: Relating supramolecular structures to cellular pathways. Expert Opin. Biol. Ther. 2005, 5, 33–53.
- Bielke, W.; Erbacher, C. (Eds.) Nucleic Acid Transfection; Springer: Berlin, Germany, 2010.
- Dan, N.; Danino, D. Structure and kinetics of lipid–nucleic acid complexes. Adv. Colloid Interface Sci. 2014, 205, 230–239.
- Ewert, K.K.; Zidovska, A.; Ahmad, A.; Bouxsein, N.F.; Evans, H.M.; McAllister, C.S.; Samuel, C.E.; Safinya, C.R. Cationic Liposome–Nucleic Acid Complexes for Gene Delivery and Silencing: Pathways and Mechanisms for Plasmid DNA and siRNA. Top. Curr. Chem. 2010, 296, 191–226.
- Safinya, C.R.; Ewert, K.K.; Majzoub, R.N.; Leal, C. Cationic liposome-nucleic acid complexes for gene delivery and gene silencing. New J. Chem. 2014, 38, 5164–5172.
- Guo, X.; Huang, L. Recent Advances in Nonviral Vectors for Gene Delivery. Acc. Chem. Res. 2012, 45, 971–979.
- Huang, L.; Hung, M.C.; Wagner, E. (Eds.) Non-Viral Vectors for Gene Therapy, 2nd ed.; Elsevier; Academic Press: San Diego, CA, USA, 2005.
- Huang, L.; Hung, M.-C.; Wagner, E. (Eds.) Nonviral Vectors for Gene Therapy; Academic Press: San Diego, CA, USA, 1999.
- Huang, L.; Liu, D.; Wagner, E. (Eds.) Nonviral Vectors for Gene Therapy: Lipid- and Polymer-Based Gene Transfer; Academic Press: New York, NY, USA, 2014.
- Lin, P.J.C.; Tam, Y.Y.C.; Hafez, I.; Sandhu, A.; Chen, S.; Ciufolini, M.A.; Nabi, I.R.; Cullis, P.R. Influence of cationic lipid composition on uptake and intracellular processing of lipid nanoparticle formulations of siRNA. Nanomedicine 2013, 9, 233–246.
- Felgner, P.L.; Gadek, T.R.; Holm, M.; Roman, R.; Chan, H.W.; Wenz, M.; Northrop, J.P.; Ringold, G.M.; Danielsen, M. Lipofection: A highly efficient, lipid-mediated DNA-transfection procedure. Proc. Natl. Acad. Sci. USA 1987, 84, 7413–7417.
- Jacob N. Israelachvili; D. John Mitchell; Barry W. Ninham; Theory of self-assembly of hydrocarbon amphiphiles into micelles and bilayers. J. Chem. Soc., Faraday Trans. 2 1975, 72, 1525-1568, 10.1039/f29767201525.
- Jacob N. Israelachvili; D.John Mitchell; Barry Ninham; Theory of self-assembly of lipid bilayers and vesicles. Biochimica et Biophysica Acta (BBA) - Biomembranes 1977, 470, 185-201, 10.1016/0005-2736(77)90099-2.
- Pieter R. Cullis; Michael J. Hope; Colin P.S. Tilcock; Lipid polymorphism and the roles of lipids in membranes. Chemistry and Physics of Lipids 1986, 40, 127-144, 10.1016/0009-3084(86)90067-8.
- Colin P.S. Tilcock; Lipid polymorphism. Chemistry and Physics of Lipids 1986, 40, 109-125, 10.1016/0009-3084(86)90066-6.
- W Helfrich; Elastic Properties of Lipid Bilayers: Theory and Possible Experiments. Zeitschrift für Naturforschung C 1973, 28, 693-703, 10.1515/znc-1973-11-1209.
- Lipowsky, R.; Sackmann, E., Eds.:. Structure and Dynamics of Membranes; Elsevier: Amsterdam, 1995; pp. 1.
- Safran, S. A.. Statistical thermodynamics of surfaces, interfaces, and membranes; Addison-Wesley: Reading, 1994; pp. 1.
- 105. Landau, L.D.; Lifshitz, E.M. . Theory of Elasticity; Volume 7 of Course of Theoretical Physics; Pergamon Press: Oxford, 1970; pp. 1.
- Igal Szleifer; Diego Kramer; Avinoam Ben-Shaul; Didier Roux; William M. Gelbart; Curvature Elasticity of Pure and Mixed Surfactant Films. Physical Review Letters 1988, 60, 1966-1969, 10.1103/physrevlett.60.1966.
- John Seddon; Structure of the inverted hexagonal (HII) phase, and non-lamellar phase transitions of lipids. Biochimica et Biophysica Acta (BBA) - Reviews on Biomembranes 1990, 1031, 1-69, 10.1016/0304-4157(90)90002-t.
- Sol M. Gruner; Stability of lyotropic phases with curved interfaces. The Journal of Physical Chemistry 1989, 93, 7562-7570, 10.1021/j100359a011.
- Yuhong Zhen; Kai K. Ewert; William S. Fisher; Victoria M. Steffes; Youli Li; Cyrus R. Safinya; Paclitaxel loading in cationic liposome vectors is enhanced by replacement of oleoyl with linoleoyl tails with distinct lipid shapes. Scientific Reports 2021, 11, 1-14, 10.1038/s41598-021-86484-9.
- James Heyes; Lorne Palmer; Kaz Bremner; Ian MacLachlan; Cationic lipid saturation influences intracellular delivery of encapsulated nucleic acids. Journal of Controlled Release 2005, 107, 276-287, 10.1016/j.jconrel.2005.06.014.
- J. P. Behr; B. Demeneix; J. P. Loeffler; J. Perez-Mutul; Efficient gene transfer into mammalian primary endocrine cells with lipopolyamine-coated DNA.. Proceedings of the National Academy of Sciences 1989, 86, 6982-6986, 10.1073/pnas.86.18.6982.
- Kai Ewert; Ayesha Ahmad; Heather M. Evans; Hans-Werner Schmidt; Cyrus R. Safinya; Efficient Synthesis and Cell-Transfection Properties of a New Multivalent Cationic Lipid for Nonviral Gene Delivery. Journal of Medicinal Chemistry 2002, 45, 5023-5029, 10.1021/jm020233w.
- Alexandra Zidovska; Kai Ewert; Joel Quispe; Bridget Carragher; Clinton S. Potter; Cyrus R. Safinya; Block liposome and nanotube formation is a general phenomenon of two-component membranes containing multivalent lipids. Soft Matter 2011, 7, 8363-8369, 10.1039/c1sm05481c.
- Victoria Marie Steffes; Zhening Zhang; Scott Macdonald; John Crowe; Kai K. Ewert; Bridget Carragher; Clinton S. Potter; Cyrus R. Safinya; PEGylation of Paclitaxel-Loaded Cationic Liposomes Drives Steric Stabilization of Bicelles and Vesicles thereby Enhancing Delivery and Cytotoxicity to Human Cancer Cells. ACS Applied Materials & Interfaces 2019, 12, 151-162, 10.1021/acsami.9b16150.
- Markus Johnsson; Katarina Edwards; Phase Behavior and Aggregate Structure in Mixtures of Dioleoylphosphatidylethanolamine and Poly(Ethylene Glycol)-Lipids. Biophysical Journal 2001, 80, 313-323, 10.1016/s0006-3495(01)76016-x.
- Maria C. Sandström; Emma Johansson; Katarina Edwards; Structure of Mixed Micelles Formed in PEG-Lipid/Lipid Dispersions. Langmuir 2007, 23, 4192-4198, 10.1021/la063501s.
- Zidovska, A.; Evans, H.M.; Ewert, K.K.; Quispe, J.; Carragher, B.; Potter, C.S.; Safinya, C.R. Liquid crystalline phases of dendritic lipid-DNA self-assemblies: Lamellar, hexagonal, and DNA bundles. J. Phys. Chem. B 2009, 113, 3694–3703.
- David P. Siegel; The Modified Stalk Mechanism of Lamellar/Inverted Phase Transitions and Its Implications for Membrane Fusion. Biophysical Journal 1999, 76, 291-313, 10.1016/s0006-3495(99)77197-3.
- G Porte; Lamellar phases and disordered phases of fluid bilayer membranes. Journal of Physics: Condensed Matter 1992, 4, 8649-8670, 10.1088/0953-8984/4/45/002.
- Lin Yang; Huey W. Huang; Observation of a Membrane Fusion Intermediate Structure. Science 2002, 297, 1877-1879, 10.1126/science.1074354.
- Gerald S. Manning; Limiting Laws and Counterion Condensation in Polyelectrolyte Solutions I. Colligative Properties. The Journal of Chemical Physics 1969, 51, 924-933, 10.1063/1.1672157.
- Alexandra S. Piotrowski-Daspit; Amy C. Kauffman; Laura G. Bracaglia; W. Mark Saltzman; Polymeric vehicles for nucleic acid delivery. Advanced Drug Delivery Reviews 2020, 156, 119-132, 10.1016/j.addr.2020.06.014.
- Brigitte Sternberg; Frank L. Sorgi; Leaf Huang; New structures in complex formation between DNA and cationic liposomes visualized by freeze-fracture electron microscopy. FEBS Letters 1994, 356, 361-366, 10.1016/0014-5793(94)01315-2.
- Joachim O. Rädler; Ilya Koltover; Tim Salditt; Cyrus R. Safinya; Structure of DNA-Cationic Liposome Complexes: DNA Intercalation in Multilamellar Membranes in Distinct Interhelical Packing Regimes. Science 1997, 275, 810-814, 10.1126/science.275.5301.810.
- Ilya Koltover; Tim Salditt; Joachim O. Rädler; Cyrus R. Safinya; An Inverted Hexagonal Phase of Cationic Liposome-DNA Complexes Related to DNA Release and Delivery. Science 1998, 281, 78-81, 10.1126/science.281.5373.78.
- Kai K. Ewert; Heather M. Evans; Alexandra Zidovska; Nathan F. Bouxsein; Ayesha Ahmad; Cyrus R. Safinya; A Columnar Phase of Dendritic Lipid−Based Cationic Liposome−DNA Complexes for Gene Delivery: Hexagonally Ordered Cylindrical Micelles Embedded in a DNA Honeycomb Lattice. Journal of the American Chemical Society 2006, 128, 3998-4006, 10.1021/ja055907h.
- Cecília Leal; Nathan F. Bouxsein; Kai K. Ewert; Cyrus R. Safinya; Highly Efficient Gene Silencing Activity of siRNA Embedded in a Nanostructured Gyroid Cubic Lipid Matrix. Journal of the American Chemical Society 2010, 132, 16841-16847, 10.1021/ja1059763.
- Danilo D. Lasic; Helmut Strey; Mark C. A. Stuart; Rudolf Podgornik; Peter M. Frederik; The Structure of DNA−Liposome Complexes. Journal of the American Chemical Society 1996, 119, 832-833, 10.1021/ja962713g.
- Stefan Huebner; Bronwyn Jean Battersby; Rudo Grimm; Gregor Cevc; Lipid-DNA Complex Formation: Reorganization and Rupture of Lipid Vesicles in the Presence of DNA As Observed by Cryoelectron Microscopy. Biophysical Journal 1999, 76, 3158-3166, 10.1016/s0006-3495(99)77467-9.
- Ramsey N. Majzoub; Kai Ewert; Erica L. Jacovetty; Bridget Carragher; Clinton S. Potter; Yichen Li; Cyrus R. Safinya; Patterned Threadlike Micelles and DNA-Tethered Nanoparticles: A Structural Study of PEGylated Cationic Liposome–DNA Assemblies. Langmuir 2015, 31, 7073-7083, 10.1021/acs.langmuir.5b00993.
- Nathan F. Bouxsein; Christopher S. McAllister; Kai K. Ewert; Charles E. Samuel; Cyrus Safinya; Structure and Gene Silencing Activities of Monovalent and Pentavalent Cationic Lipid Vectors Complexed with siRNA. Biochemistry 2007, 46, 4785-4792, 10.1021/bi062138l.
- Antje Ziller; Sara S. Nogueira; Eva Hühn; Sergio S. Funari; Gerald Brezesinski; Hermann Hartmann; Ugur Sahin; Heinrich Haas; Peter Langguth; Incorporation of mRNA in Lamellar Lipid Matrices for Parenteral Administration. Molecular Pharmaceutics 2018, 15, 642-651, 10.1021/acs.molpharmaceut.7b01022.
- F. Artzner; R. Zantl; G. Rapp; J. O. Rädler; Observation of a Rectangular Columnar Phase in Condensed Lamellar Cationic Lipid-DNA Complexes. Physical Review Letters 1998, 81, 5015-5018, 10.1103/physrevlett.81.5015.
- Rumiana Koynova; Robert C. MacDonald; Columnar DNA Superlattices in Lamellar o-Ethylphosphatidylcholine Lipoplexes: Mechanism of the Gel-Liquid Crystalline Lipid Phase Transition. Nano Letters 2004, 4, 1475-1479, 10.1021/nl049191k.
- § Jennifer J. McManus; ‡ And Joachim O. Rädler; † Kenneth A. Dawson; Observation of a Rectangular Columnar Phase in a DNA−Calcium−Zwitterionic Lipid Complex. Journal of the American Chemical Society 2004, 126, 15966-15967, 10.1021/ja046105+.
- Koltover, I.; Salditt, T.; Rädler, J.O.; Safinya, C.R. An inverted hexagonal phase of cationic liposome-DNA complexes related to DNA release and delivery. Science 1998, 281, 78–81.
- Alison J. Lin; Nelle L. Slack; Ayesha Ahmad; Cyril X. George; Charles E. Samuel; Cyrus R. Safinya; Three-Dimensional Imaging of Lipid Gene-Carriers: Membrane Charge Density Controls Universal Transfection Behavior in Lamellar Cationic Liposome-DNA Complexes. Biophysical Journal 2003, 84, 3307-3316, 10.1016/s0006-3495(03)70055-1.
- Ewert, K.K.; Evans, H.M.; Zidovska, A.; Bouxsein, N.F.; Ahmad, A.; Safinya, C.R. A columnar phase of dendritic lipid-based cationic liposome-DNA complexes for gene delivery: Hexagonally ordered cylindrical micelles embedded in a DNA honeycomb lattice. J. Am. Chem. Soc. 2006, 128, 3998–4006.
- Chandrashekhar V. Kulkarni; Wolfgang Wachter; Guillermo Iglesias-Salto; Sandra Engelskirchen; Silvia Ahualli; Monoolein: a magic lipid?. Physical Chemistry Chemical Physics 2010, 13, 3004-3021, 10.1039/c0cp01539c.
- Cecília Leal; Kai K. Ewert; Rahau S. Shirazi; Nathan F. Bouxsein; Cyrus R. Safinya; Nanogyroids Incorporating Multivalent Lipids: Enhanced Membrane Charge Density and Pore Forming Ability for Gene Silencing. Langmuir 2011, 27, 7691-7697, 10.1021/la200679x.
- Minjee Kang; Hojun Kim; Cecilia Leal; Self-organization of nucleic acids in lipid constructs. Current Opinion in Colloid & Interface Science 2016, 26, 58-65, 10.1016/j.cocis.2016.09.006.
- María Martínez-Negro; Krishan Kumar; Ana L. Barrán-Berdón; Sougata Datta; Paturu Kondaiah; Elena Junquera; Santanu Bhattacharya; Emilio Aicart; Efficient Cellular Knockdown Mediated by siRNA Nanovectors of Gemini Cationic Lipids Having Delocalizable Headgroups and Oligo-Oxyethylene Spacers. ACS Applied Materials & Interfaces 2016, 8, 22113-22126, 10.1021/acsami.6b08823.
- Hojun Kim; Cecilia Leal; Cuboplexes: Topologically Active siRNA Delivery. ACS Nano 2015, 9, 10214-10226, 10.1021/acsnano.5b03902.
- Hojun Kim; Jaeuk Sung; YunJu Chang; Alana Alfeche; Cecilia Leal; Microfluidics Synthesis of Gene Silencing Cubosomes. ACS Nano 2018, 12, 9196-9205, 10.1021/acsnano.8b03770.
- Cecília Leal; Kai Ewert; Nathan F. Bouxsein; Rahau S. Shirazi; Youli Li; Cyrus R. Safinya; Stacking of short DNA induces the gyroid cubic-to-inverted hexagonal phase transition in lipid–DNA complexes. Soft Matter 2012, 9, 795-804, 10.1039/c2sm27018h.
- Campbell, R.B.; Ying, B.; Kuesters, G.M.; Hemphill, R. Fighting cancer: From the bench to bedside using second generation cationic liposomal therapeutics. J. Pharm. Sci. 2009, 98, 411–429.
- Strieth, S.; Eichhorn, M.E.; Sauer, B.; Schulze, B.; Teifel, M.; Michaelis, U.; Dellian, M. Neovascular targeting chemotherapy: Encapsulation of paclitaxel in cationic liposomes impairs functional tumor microvasculature. Int. J. Cancer 2004, 110, 117–124.
- Strieth, S.; Nussbaum, C.F.; Eichhorn, M.E.; Fuhrmann, M.; Teifel, M.; Michaelis, U.; Berghaus, A.; Dellian, M. Tumor-selective vessel occlusions by platelets after vascular targeting chemotherapy using paclitaxel encapsulated in cationic liposomes. Int. J. Cancer 2008, 122, 452–460.
- Schmitt-Sody, M.; Strieth, S.; Krasnici, S.; Sauer, B.; Schulze, B.; Teifel, M.; Michaelis, U.; Naujoks, K.; Dellian, M. Neovascular Targeting Therapy: Paclitaxel Encapsulated in Cationic Liposomes Improves Antitumoral Efficacy. Clin. Cancer Res. 2003, 9, 2335–2341.
- Kunstfeld, R.; Wickenhauser, G.; Michaelis, U.; Teifel, M.; Umek, W.; Naujoks, K.; Wolff, K.; Petzelbauer, P. Paclitaxel Encapsulated in Cationic Liposomes Diminishes Tumor Angiogenesis and Melanoma Growth in a “Humanized” SCID Mouse Model. J. Investig. Dermatol. 2003, 120, 476–482.
- Fasol, U.; Frost, A.; Büchert, M.; Arends, J.; Fiedler, U.; Scharr, D.; Scheuenpflug, J.; Mross, K. Vascular and pharmacokinetic effects of EndoTAG-1 in patients with advanced cancer and liver metastasis. Ann. Oncol. 2012, 23, 1030–1036.
- Koudelka, Š.; Turánek, J. Liposomal paclitaxel formulations. J. Control. Release 2012, 163, 322–334.
- Wani, M.C.; Taylor, H.L.; Wall, M.E.; Coggon, P.; McPhail, A.T. Plant antitumor agents. VI. Isolation and structure of taxol, a novel antileukemic and antitumor agent from Taxus brevifolia. J. Am. Chem. Soc. 1971, 93, 2325–2327.
- Jordan, M.A.; Wilson, L. Microtubules as a target for anticancer drugs. Nat. Rev. Cancer 2004, 4, 253–265.
- Weaver, B.A. How Taxol/paclitaxel kills cancer cells. Mol. Biol. Cell 2014, 25, 2677–2681.
- Rowinsky, E.K.; Donehower, R.C. Paclitaxel (Taxol). N. Engl. J. Med. 1995, 332, 1004–1014.
- Markman, M.; Mekhail, T.M. Paclitaxel in cancer therapy. Expert Opin. Pharmacother. 2002, 3, 755–766.
- Ramalingam, S.; Belani, C.P. Paclitaxel for non-small cell lung cancer. Expert Opin. Pharmacother. 2004, 5, 1771–1780.
- Hironaka, S.; Zenda, S.; Boku, N.; Fukutomi, A.; Yoshino, T.; Onozawa, Y. Weekly paclitaxel as second-line chemotherapy for advanced or recurrent gastric cancer. Gastric Cancer 2006, 9, 14–18.
- Sakamoto, J.; Matsui, T.; Kodera, Y. Paclitaxel chemotherapy for the treatment of gastric cancer. Gastric Cancer 2009, 12, 69–78.
- Moxley, K.M.; McMeekin, D.S. Endometrial Carcinoma: A Review of Chemotherapy, Drug Resistance, and the Search for New Agents. Oncologist 2010, 15, 1026–1033.
- Teo, P.Y.; Cheng, W.; Hedrick, J.L.; Yang, Y.Y. Co-delivery of drugs and plasmid DNA for cancer therapy. Adv. Drug Deliv. Rev. 2016, 98, 41–63.
- Dorr, R.T. Pharmacology and Toxicology of Cremophor EL Diluent. Ann. Pharmacother. 1994, 28, S11–S14.
- Weiss, R.B.; Donehower, R.C.; Wiernik, P.H.; Ohnuma, T.; Gralla, R.J.; Trump, D.L.; Jr, J.R.B.; Echo, D.A.V.; Hoff, D.D.V.; Leyland-Jones, B. Hypersensitivity reactions from taxol. J. Clin. Oncol. 1990, 8, 1263–1268.
- Gelderblom, H.; Verweij, J.; Nooter, K.; Sparreboom, A. Cremophor EL: The drawbacks and advantages of vehicle selection for drug formulation. Eur. J. Cancer 2001, 37, 1590–1598.
- Sofias, A.M.; Dunne, M.; Storm, G.; Allen, C. The battle of “nano” paclitaxel. Adv. Drug Deliv. Rev. 2017, 122, 20–30.
- Hong, S.-S.; Choi, J.Y.; Kim, J.O.; Lee, M.-K.; Kim, S.H.; Lim, S.-J. Development of paclitaxel-loaded liposomal nanocarrier stabilized by triglyceride incorporation. Int. J. Nanomed. 2016, 11, 4465–4477.
- Zhou, R.; Mazurchuk, R.V.; Tamburlin, J.H.; Harrold, J.M.; Mager, D.E.; Straubinger, R.M. Differential Pharmacodynamic Effects of Paclitaxel Formulations in an Intracranial Rat Brain Tumor Model. J. Pharmacol. Exp. Ther. 2010, 332, 479–488.
- Ait-Oudhia, S.; Mager, D.E.; Straubinger, R.M. Application of Pharmacokinetic and Pharmacodynamic Analysis to the Development of Liposomal Formulations for Oncology. Pharmaceutics 2014, 6, 137–174.
- Zhang, J.A.; Anyarambhatla, G.; Ma, L.; Ugwu, S.; Xuan, T.; Sardone, T.; Ahmad, I. Development and characterization of a novel Cremophor® EL free liposome-based paclitaxel (LEP-ETU) formulation. Eur. J. Pharm. Biopharm. 2005, 59, 177–187.
- Dellian, M.; Yuan, F.; Trubetskoy, V.S.; Torchilin, V.P.; Jain, R.K. Vascular permeability in a human tumour xenograft: Molecular charge dependence. Br. J. Cancer 2000, 82, 1513–1518.
- Eichhorn, M.E.; Ischenko, I.; Luedemann, S.; Strieth, S.; Papyan, A.; Werner, A.; Bohnenkamp, H.; Guenzi, E.; Preissler, G.; Michaelis, U.; et al. Vascular targeting by EndoTAG™-1 enhances therapeutic efficacy of conventional chemotherapy in lung and pancreatic cancer. Int. J. Cancer 2010, 126, 1235–1245.
- Koudelka, Š.; Turánek-Knötigová, P.; MaŠek, J.; Korvasová, Z.; Škrabalová, M.; Plocková, J.; Bartheldyová, E.; Turánek, J. Liposomes with high encapsulation capacity for paclitaxel: Preparation, characterisation and in vivo anticancer effect. J. Pharm. Sci. 2010, 99, 2309–2319.
- Koudelka, Š.; Turánek-Knötigová, P.; MaŠek, J.; Korvasová, Z.; Škrabalová, M.; Plocková, J.; Bartheldyová, E.; Turánek, J. Liposomes with high encapsulation capacity for paclitaxel: Preparation, characterisation and in vivo anticancer effect. J. Pharm. Sci. 2010, 99, 2309–2319.
- Koudelka, Š.; Turánek-Knötigová, P.; MaŠek, J.; Korvasová, Z.; Škrabalová, M.; Plocková, J.; Bartheldyová, E.; Turánek, J. Liposomes with high encapsulation capacity for paclitaxel: Preparation, characterisation and in vivo anticancer effect. J. Pharm. Sci. 2010, 99, 2309–2319.
- Jacob N. Israelachvili; D.John Mitchell; Barry Ninham; Theory of self-assembly of lipid bilayers and vesicles. Biochimica et Biophysica Acta (BBA) - Biomembranes 1977, 470, 185-201, 10.1016/0005-2736(77)90099-2.
- Pieter R. Cullis; Michael J. Hope; Colin P.S. Tilcock; Lipid polymorphism and the roles of lipids in membranes. Chemistry and Physics of Lipids 1986, 40, 127-144, 10.1016/0009-3084(86)90067-8.