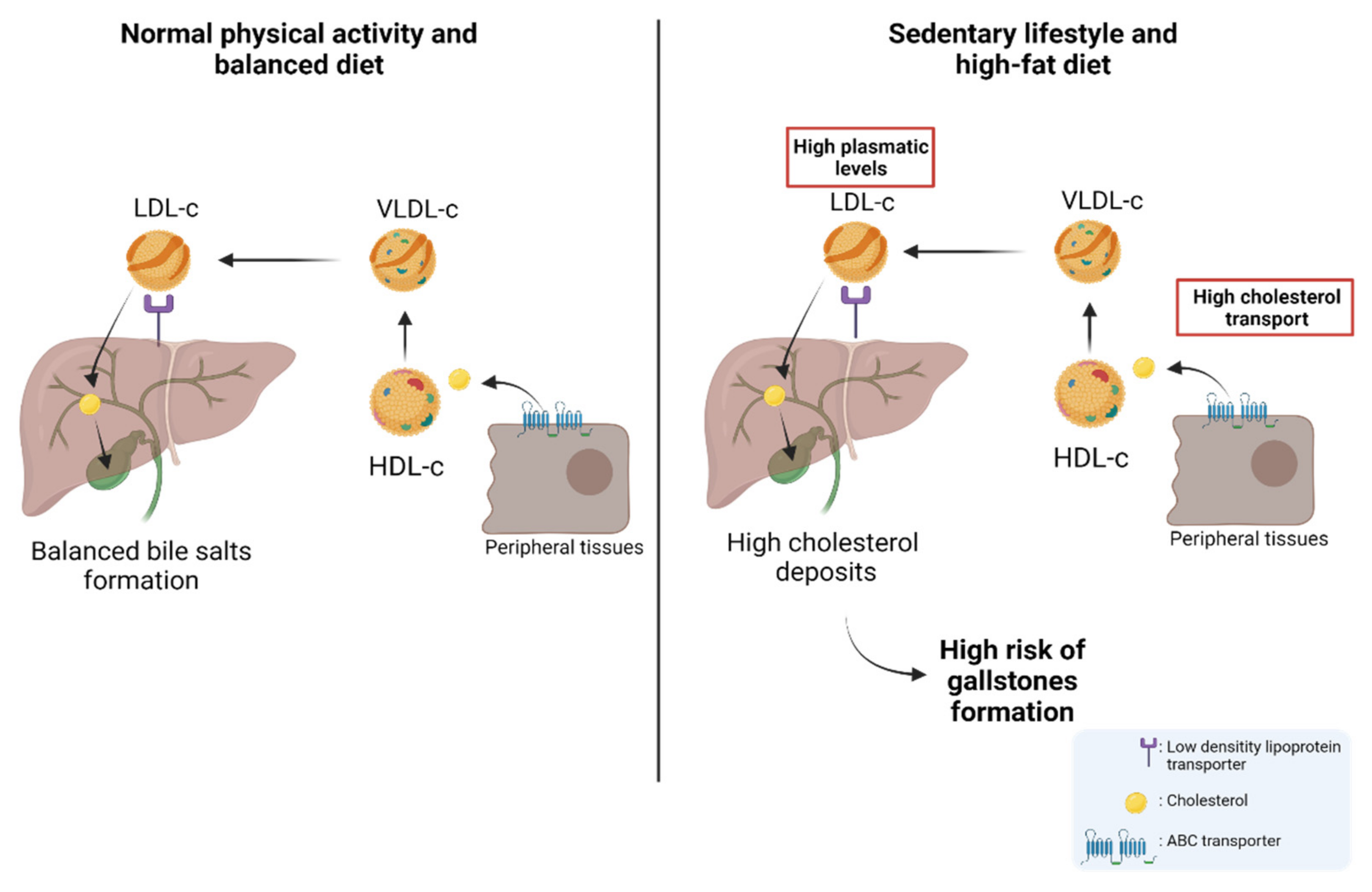
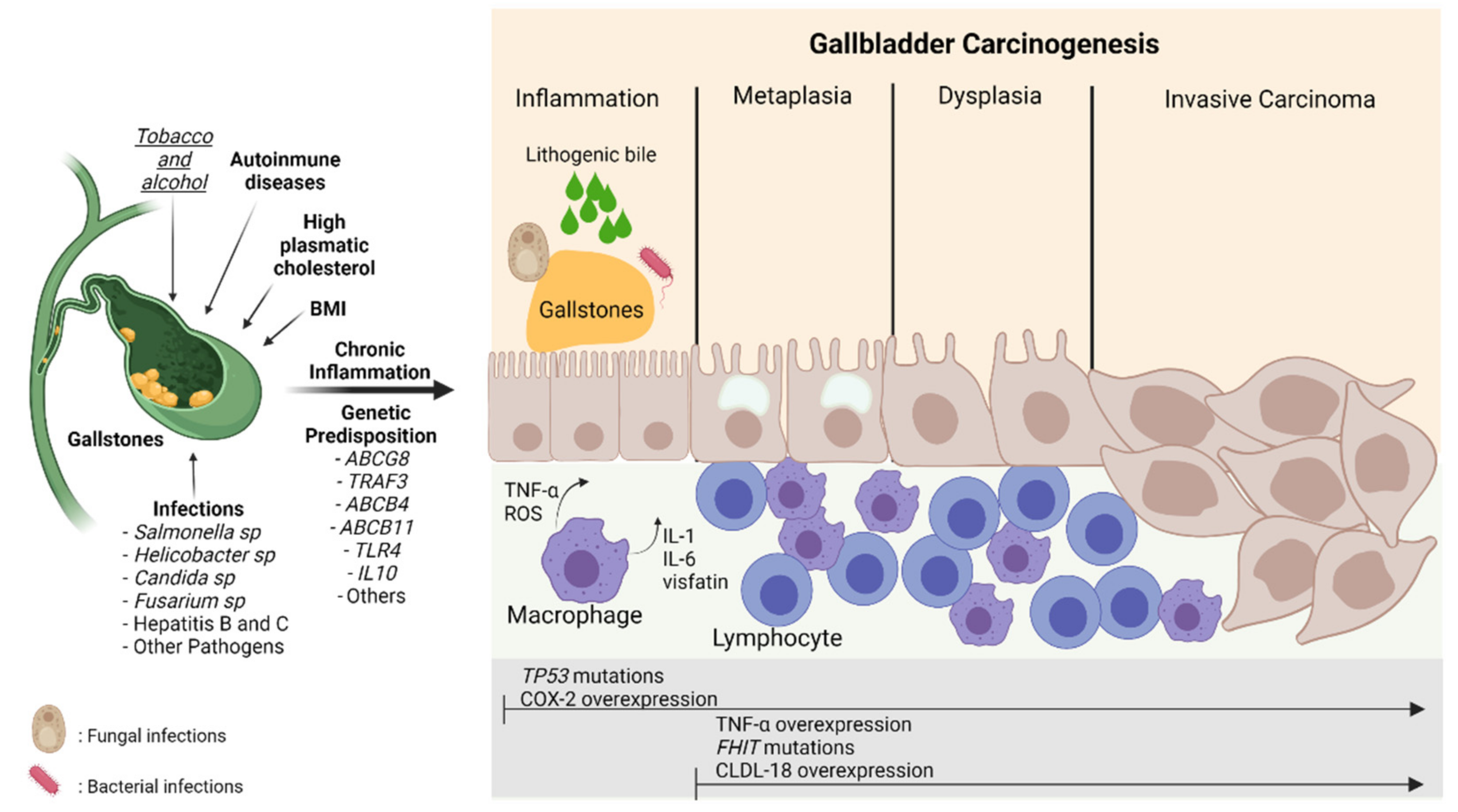
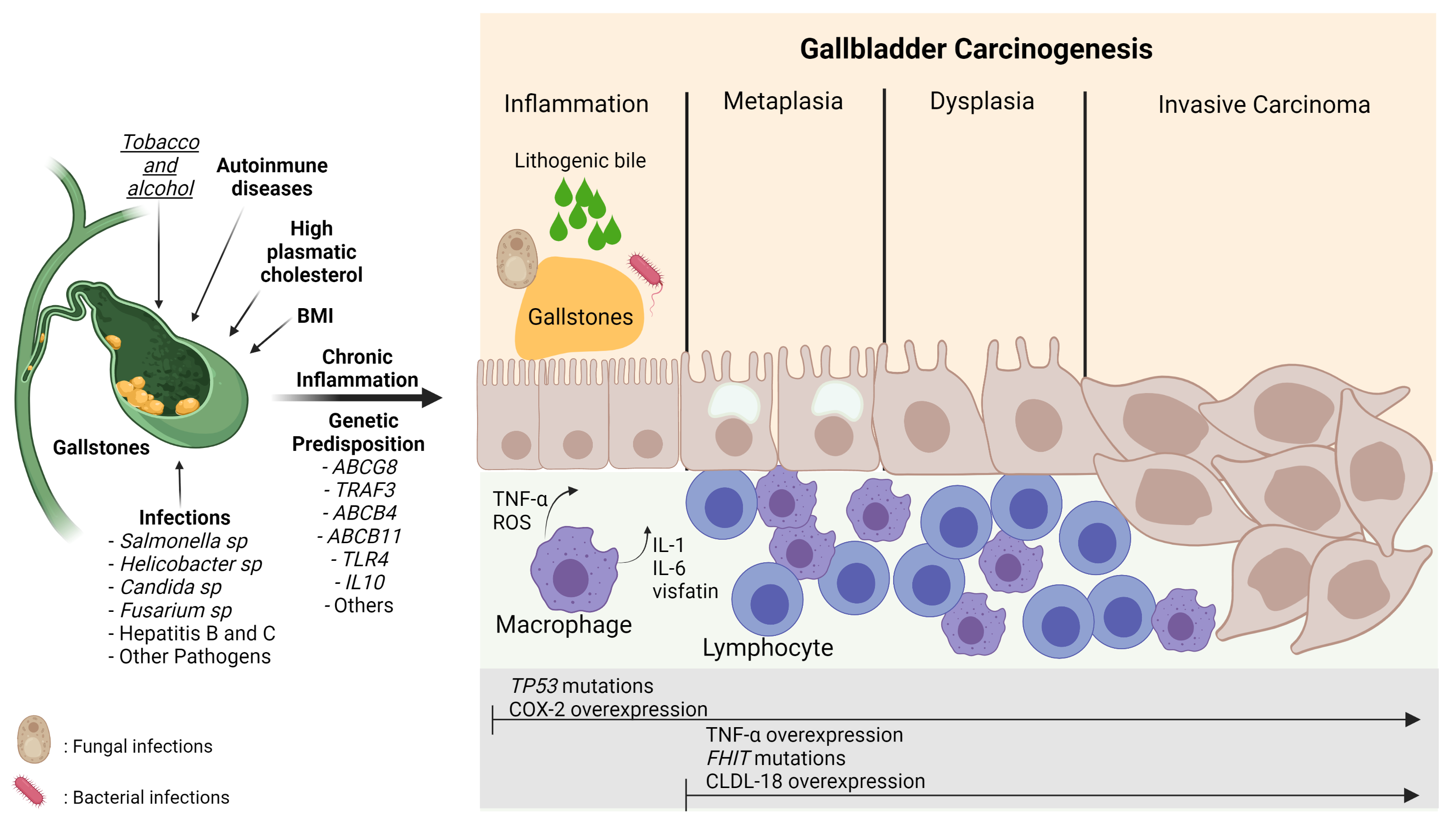
Gallbladder cancer (GBC) is an aggressive neoplasm that in an early stage is generally asymptomatic and, in most cases, is diagnosed in advanced stages with a very low life expectancy because there is no curative treatment. Therefore, understanding the early carcinogenic mechanisms of this pathology is crucial to proposing preventive strategies for this cancer. The main risk factor is the presence of gallstones, which are associated with some environmental factors such as a sedentary lifestyle and a high-fat diet. Other risk factors such as autoimmune disorders and bacterial, parasitic and fungal infections have also been described. All these factors can generate a long-term inflammatory state characterized by the persistent activation of the immune system, the frequent release of pro-inflammatory cytokines, and the constant production of reactive oxygen species that result in a chronic damage/repair cycle, subsequently inducing the loss of the normal architecture of the gallbladder mucosa that leads to the development of GBC.
- gallbladder cancer
- risk factors
- carcinogenesis
1. Introduction
2. Nutritional and Lifestyle Aspects That Increase Susceptibility to Gallbladder Carcinogenesis
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249.
- Hundal, R.; Shaffer, E.A. Gallbladder cancer: Epidemiology and outcome. Clin Epidemiol 2014, 6, 99–109.
- Alvi, A.R.; Siddiqui, N.A.; Zafar, H. Risk factors of gallbladder cancer in Karachi-a case-control study. World J. Surg. Oncol. 2011, 9, 164.
- Roa, I.; Ibacache, G.; Roa, J.; Araya, J.; de Aretxabala, X.; Muñoz, S. Gallstones and gallbladder cancer-volume and weight of gallstones are associated with gallbladder cancer: A case-control study. J. Surg. Oncol. 2006, 93, 624–628.
- Hsing, A.W.; Gao, Y.-T.; Han, T.-Q.; Rashid, A.; Sakoda, L.C.; Wang, B.-S.; Shen, M.-C.; Zhang, B.-H.; Niwa, S.; Chen, J.; et al. Gallstones and the risk of biliary tract cancer: A population-based study in China. Br. J. Cancer 2007, 97, 1577–1582.
- Espinoza, J.A.; Bizama, C.; García, P.; Ferreccio, C.; Javle, M.; Miquel, J.F.; Koshiol, J.; Roa, J.C. The inflammatory inception of gallbladder cancer. Biochim. Et Biophys. Acta (BBA)-Bioenerg. 2016, 1865, 245–254.
- Stokes, C.S.; Krawczyk, M.; Lammert, F. Gallstones: Environment, Lifestyle and Genes. Dig. Dis. 2011, 29, 191–201.
- Bouvard, V.; Baan, R.; Straif, K.; Grosse, Y.; Secretan, B.; El Ghissassi, F.; Benbrahim-Tallaa, L.; Guha, N.; Freeman, C.; Galichet, L.; et al. A review of human carcinogens—Part B: Biological agents. Lancet Oncol. 2009, 10, 321–322.
- Nagaraja, V.; Eslick, G.D. Systematic review with meta-analysis: The relationship between chronicSalmonella typhicarrier status and gall-bladder cancer. Aliment. Pharmacol. Ther. 2014, 39, 745–750.
- Zhu, Q.; Sun, X.; Ji, X.; Zhu, L.; Xu, J.; Wang, C.; Zhang, C.; Xue, F.; Liu, Y. The association between gallstones and metabolic syndrome in urban Han Chinese: A longitudinal cohort study. Sci. Rep. 2016, 6, 29937.
- van Erp, L.W.; Cunningham, M.; Narasimman, M.; Ali, H.A.; Jhaveri, K.; Drenth, J.P.H.; Janssen, H.L.A.; Levy, C.; Hirschfield, G.M.; Hansen, B.E.; et al. Risk of gallbladder cancer in patients with primary sclerosing cholangitis and radiographically detected gallbladder polyps. Liver Int. 2020, 40, 382–392.
- Deng, Z.; Xuan, Y.; Li, X.; Crawford, W.J.; Yuan, Z.; Chen, Z.; Brooks, A.; Song, Y.; Wang, H.; Liang, X.; et al. Effect of metabolic syndrome components on the risk of malignancy in patients with gallbladder lesions. J. Cancer 2021, 12, 1531–1537.
- Gu, J.; Yan, S.; Wang, B.; Shen, F.; Cao, H.; Fan, J.; Wang, Y. Type 2 diabetes mellitus and risk of gallbladder cancer: A systematic review and meta-analysis of observational studies. Diabetes/Metab. Res. Rev. 2015, 32, 63–72.
- Wenbin, D.; Zhuo, C.; Zhibing, M.; Chen, Z.; Ruifan, Y.; Jie, J.; Cheng, Q.; Zhenming, G. The effect of smoking on the risk of gallbladder cancer: A meta-analysis of observational studies. Eur. J. Gastroenterol. Hepatol. 2013, 25, 373–379.
- Yagyu, K.; Kikuchi, S.; Obata, Y.; Lin, Y.; Ishibashi, T.; Kurosawa, M.; Inaba, Y.; Tamakoshi, A.; JACC Study Group. Cigarette smoking, alcohol drinking and the risk of gallbladder cancer death: A prospective cohort study in Japan. Int. J. Cancer 2008, 122, 924–929.
- McGee, E.E.; Jackson, S.S.; Petrick, J.L.; Van Dyke, A.L.; Adami, H.-O.; Albanes, D.; Andreotti, G.; Beane-Freeman, L.E.; De Gonzalez, A.B.; Buring, J.E.; et al. Smoking, Alcohol, and Biliary Tract Cancer Risk: A Pooling Project of 26 Prospective Studies. J. Natl. Cancer Inst. 2019, 111, 1263–1278.
- Pandey, M.; Shukla, V.K. Diet and gallbladder cancer: A case-control study. Eur. J. Cancer Prev. 2002, 11, 365–368.
- Di Ciaula, A.; Portincasa, P. Recent advances in understanding and managing cholesterol gallstones. F1000Res 2018, 7.
- Skoumas, J.; Pitsavos, C.; Panagiotakos, D.B.; Chrysohoou, C.; Zeimbekis, A.; Papaioannou, I.; Toutouza, M.; Toutouzas, P.; Stefanadis, C. Physical activity, high density lipoprotein cholesterol and other lipids levels, in men and women from the ATTICA study. Lipids Health Dis. 2003, 2, 3.
- Renehan, A.G.; Tyson, M.; Egger, M.; Heller, R.F.; Zwahlen, M. Body-mass index and incidence of cancer: A systematic review and meta-analysis of prospective observational studies. Lancet 2008, 371, 569–578.
- Hsing, A.W.; Sakoda, L.C.; Rashid, A.; Chen, J.; Shen, M.C.; Han, T.Q.; Wang, B.S.; Gao, Y.T. Body size and the risk of biliary tract cancer: A population-based study in China. Br. J. Cancer 2008, 99, 811–815.
- Tan, W.; Gao, M.; Liu, N.; Zhang, G.; Xu, T.; Cui, W. Body Mass Index and Risk of Gallbladder Cancer: Systematic Review and Meta-Analysis of Observational Studies. Nutrients 2015, 7, 8321–8334.
- Everson, G.T.; McKinley, C.; Kern, F. Mechanisms of gallstone formation in women. Effects of exogenous estrogen (Premarin) and dietary cholesterol on hepatic lipid metabolism. J. Clin. Invest. 1991, 87, 237–246.
- Barahona Ponce, C.; Scherer, D.; Brinster, R.; Boekstegers, F.; Marcelain, K.; Gárate-Calderón, V.; Müller, B.; de Toro, G.; Retamales, J.; Barajas, O.; et al. Gallstones, Body Mass Index, C-Reactive Protein, and Gallbladder Cancer: Mendelian Randomization Analysis of Chilean and European Genotype Data. Hepatology 2021, 73, 1783–1796.
- Pinto Pereira, S.M.; Ki, M.; Power, C. Sedentary behaviour and biomarkers for cardiovascular disease and diabetes in mid-life: The role of television-viewing and sitting at work. PLoS ONE 2012, 7, e31132.
- Crichton, G.E.; Alkerwi, A. Physical activity, sedentary behavior time and lipid levels in the Observation of Cardiovascular Risk Factors in Luxembourg study. Lipids Health Dis. 2015, 14, 87.
- Larsson, S.C.; Giovannucci, E.L.; Wolk, A. Sweetened Beverage Consumption and Risk of Biliary Tract and Gallbladder Cancer in a Prospective Study. J. Natl. Cancer Inst. 2016, 108.
- Borena, W.; Edlinger, M.; Bjørge, T.; Häggström, C.; Lindkvist, B.; Nagel, G.; Engeland, A.; Stocks, T.; Strohmaier, S.; Manjer, J.; et al. A prospective study on metabolic risk factors and gallbladder cancer in the metabolic syndrome and cancer (Me-Can) collaborative study. PLoS ONE 2014, 9, e89368.
- Arcidiacono, B.; Iiritano, S.; Nocera, A.; Possidente, K.; Nevolo, M.T.; Ventura, V.; Foti, D.; Chiefari, E.; Brunetti, A. Insulin resistance and cancer risk: An overview of the pathogenetic mechanisms. Exp. Diabetes Res. 2012, 2012, 789174.
- Van Erpecum, K.J. Pathogenesis of cholesterol and pigment gallstones: An update. Clin. Res. Hepatol. Gastroenterol. 2011, 35, 281–287.
- Rebholz, C.; Krawczyk, M.; Lammert, F. Genetics of gallstone disease. Eur. J. Clin. Invest. 2018, 48, e12935.
- Ryu, S.; Chang, Y.; Yun, K.E.; Jung, H.S.; Shin, J.H.; Shin, H. Gallstones and the Risk of Gallbladder Cancer Mortality: A Cohort Study. Am. J. Gastroenterol. 2016, 111, 1476–1487.
- Stinton, L.M.; Shaffer, E.A. Epidemiology of gallbladder disease: Cholelithiasis and cancer. Gut Liver 2012, 6, 172–187.
- Rousset, X.; Vaisman, B.; Amar, M.; Sethi, A.A.; Remaley, A.T. Lecithin: Cholesterol acyltransferase--from biochemistry to role in cardiovascular disease. Curr. Opin. Endocrinol. Diabetes Obes. 2009, 16, 163–171.
- Masson, D.; Jiang, X.C.; Lagrost, L.; Tall, A.R. The role of plasma lipid transfer proteins in lipoprotein metabolism and atherogenesis. J. Lipid Res. 2009, 50, S201–S206.
- Acton, S.; Rigotti, A.; Landschulz, K.T.; Xu, S.; Hobbs, H.H.; Krieger, M. Identification of scavenger receptor SR-BI as a high density lipoprotein receptor. Science 1996, 271, 518–520.
- Barter, P.J.; Brewer, H.B.; Chapman, M.J.; Hennekens, C.H.; Rader, D.J.; Tall, A.R. Cholesteryl ester transfer protein: A novel target for raising HDL and inhibiting atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 160–167.
- Rader, D.J. Regulation of reverse cholesterol transport and clinical implications. Am. J. Cardiol. 2003, 92, 42J–49J.
- Atamanalp, S.S.; Keles, M.S.; Atamanalp, R.S.; Acemoglu, H.; Laloglu, E. The effects of serum cholesterol, LDL, and HDL levels on gallstone cholesterol concentration. Pak. J. Med. Sci. 2013, 29, 187–190.
- Wang, J.; Shen, S.; Wang, B.; Ni, X.; Liu, H.; Yu, R.; Suo, T. Serum lipid levels are the risk factors of gallbladder stones: A population-based study in China. Lipids Health Dis. 2020, 19, 50.
- Hayat, S.; Hassan, Z.; Changazi, S.H.; Zahra, A.; Noman, M.; Zain Ul Abdin, M.; Javed, H.; Ans, A.H. Comparative analysis of serum lipid profiles in patients with and without gallstones: A prospective cross-sectional study. Ann. Med. Surg. 2019, 42, 11–13.
- Reshetnyak, V.I. Concept of the pathogenesis and treatment of cholelithiasis. World J. Hepatol. 2012, 4, 18–34.
- Shrikhande, S.V.; Barreto, S.G.; Singh, S.; Udwadia, T.E.; Agarwal, A.K. Cholelithiasis in gallbladder cancer: Coincidence, cofactor, or cause! Eur. J. Surg. Oncol. 2010, 36, 514–519.
- Kuraishy, A.; Karin, M.; Grivennikov, S.I. Tumor promotion via injury- and death-induced inflammation. Immunity 2011, 35, 467–477.
- Jang, S.I.; Fang, S.; Kim, K.P.; Ko, Y.; Kim, H.; Oh, J.; Hong, G.Y.; Lee, S.Y.; Kim, J.M.; Noh, I.; et al. Combination treatment with n-3 polyunsaturated fatty acids and ursodeoxycholic acid dissolves cholesterol gallstones in mice. Sci. Rep. 2019, 9, 12740.
- Maurer, K.J.; Rao, V.P.; Ge, Z.; Rogers, A.B.; Oura, T.J.; Carey, M.C.; Fox, J.G. T-cell function is critical for murine cholesterol gallstone formation. Gastroenterology 2007, 133, 1304–1315.
- Rosa, L.; Lobos-González, L.; Muñoz-Durango, N.; García, P.; Bizama, C.; Gómez, N.; González, X.; Wichmann, I.A.; Saavedra, N.; Guevara, F.; et al. Evaluation of the chemopreventive potentials of ezetimibe and aspirin in a novel mouse model of gallbladder preneoplasia. Mol. Oncol. 2020, 14, 2834–2852.
- Lavoie, B.; Nausch, B.; Zane, E.A.; Leonard, M.R.; Balemba, O.B.; Bartoo, A.C.; Wilcox, R.; Nelson, M.T.; Carey, M.C.; Mawe, G.M. Disruption of gallbladder smooth muscle function is an early feature in the development of cholesterol gallstone disease. Neurogastroenterol. Motil. 2012, 24, e313–e324.
- Kato, S.; Fushimi, K.; Yabuki, Y.; Maru, Y.; Hasegawa, S.; Matsuura, T.; Kurotaki, D.; Suzuki, A.; Kobayashi, N.; Yoneda, M.; et al. Precision modeling of gall bladder cancer patients in mice based on orthotopic implantation of organoid-derived tumor buds. Oncogenesis 2021, 10, 33.
- Liu, Z.; Kemp, T.J.; Gao, Y.T.; Corbel, A.; McGee, E.E.; Wang, B.; Shen, M.C.; Rashid, A.; Hsing, A.W.; Hildesheim, A.; et al. Association of circulating inflammation proteins and gallstone disease. J. Gastroenterol. Hepatol. 2018, 33, 1920–1924.
- Moschen, A.R.; Kaser, A.; Enrich, B.; Mosheimer, B.; Theurl, M.; Niederegger, H.; Tilg, H. Visfatin, an adipocytokine with proinflammatory and immunomodulating properties. J. Immunol. 2007, 178, 1748–1758.
- Park, J.W.; Kim, O.H.; Lee, S.C.; Kim, K.H.; Hong, H.E.; Seo, H.; Choi, H.J.; Kim, S.J. Serum level of visfatin can reflect the severity of inflammation in patients with acute cholecystitis. Ann. Surg. Treat. Res. 2020, 99, 26–36.
- Grünhage, F.; Acalovschi, M.; Tirziu, S.; Walier, M.; Wienker, T.F.; Ciocan, A.; Mosteanu, O.; Sauerbruch, T.; Lammert, F. Increased gallstone risk in humans conferred by common variant of hepatic ATP-binding cassette transporter for cholesterol. Hepatology 2007, 46, 793–801.
- Small, D.M. Role of ABC transporters in secretion of cholesterol from liver into bile. Proc Natl Acad Sci USA 2003, 100, 4–6.
- Von Kampen, O.; Buch, S.; Nothnagel, M.; Azocar, L.; Molina, H.; Brosch, M.; Erhart, W.; von Schönfels, W.; Egberts, J.; Seeger, M.; et al. Genetic and functional identification of the likely causative variant for cholesterol gallstone disease at the ABCG5/8 lithogenic locus. Hepatology 2013, 57, 2407–2417.
- Mhatre, S.; Wang, Z.; Nagrani, R.; Badwe, R.; Chiplunkar, S.; Mittal, B.; Yadav, S.; Zhang, H.; Chung, C.C.; Patil, P.; et al. Common genetic variation and risk of gallbladder cancer in India: A case-control genome-wide association study. Lancet Oncol. 2017, 18, 535–544.
- Bustos, B.I.; Pérez-Palma, E.; Buch, S.; Azócar, L.; Riveras, E.; Ugarte, G.D.; Toliat, M.; Nürnberg, P.; Lieb, W.; Franke, A.; et al. Variants in ABCG8 and TRAF3 genes confer risk for gallstone disease in admixed Latinos with Mapuche Native American ancestry. Sci. Rep. 2019, 9, 772.
- Jackson, S.S.; Van De Wyngard, V.; Pfeiffer, R.M.; Cook, P.; Hildesheim, A.; Pinto, L.A.; Jackson, S.H.; Choi, K.; Verdugo, R.A.; Cuevas, M.; et al. Inflammatory profiles in Chilean Mapuche and non-Mapuche women with gallstones at risk of developing gallbladder cancer. Sci. Rep. 2021, 11, 3686.
- Delaunay, J.L.; Durand-Schneider, A.M.; Dossier, C.; Falguières, T.; Gautherot, J.; Davit-Spraul, A.; Aït-Slimane, T.; Housset, C.; Jacquemin, E.; Maurice, M. A functional classification of ABCB4 variations causing progressive familial intrahepatic cholestasis type 3. Hepatology 2016, 63, 1620–1631.
- Nakeeb, A.; Comuzzie, A.G.; Martin, L.; Sonnenberg, G.E.; Swartz-Basile, D.; Kissebah, A.H.; Pitt, H.A. Gallstones: Genetics versus environment. Ann. Surg. 2002, 235, 842–849.
- Rosmorduc, O.; Hermelin, B.; Boelle, P.Y.; Parc, R.; Taboury, J.; Poupon, R. ABCB4 gene mutation-associated cholelithiasis in adults. Gastroenterology 2003, 125, 452–459.
- Jansen, P.L.; Strautnieks, S.S.; Jacquemin, E.; Hadchouel, M.; Sokal, E.M.; Hooiveld, G.J.; Koning, J.H.; De Jager-Krikken, A.; Kuipers, F.; Stellaard, F.; et al. Hepatocanalicular bile salt export pump deficiency in patients with progressive familial intrahepatic cholestasis. Gastroenterology 1999, 117, 1370–1379.
- Pullinger, C.R.; Eng, C.; Salen, G.; Shefer, S.; Batta, A.K.; Erickson, S.K.; Verhagen, A.; Rivera, C.R.; Mulvihill, S.J.; Malloy, M.J.; et al. Human cholesterol 7alpha-hydroxylase (CYP7A1) deficiency has a hypercholesterolemic phenotype. J. Clin. Invest. 2002, 110, 109–117.
- Miyake, J.H.; Duong-Polk, X.T.; Taylor, J.M.; Du, E.Z.; Castellani, L.W.; Lusis, A.J.; Davis, R.A. Transgenic expression of cholesterol-7-alpha-hydroxylase prevents atherosclerosis in C57BL/6J mice. Arterioscler. Thromb. Vasc. Biol. 2002, 22, 121–126.
- Lancellotti, S.; Zaffanello, M.; Di Leo, E.; Costa, L.; Lonardo, A.; Tarugi, P. Pediatric gallstone disease in familial hypobetalipoproteinemia. J. Hepatol. 2005, 43, 188–191.
- Miller, L.J.; Holicky, E.L.; Ulrich, C.D.; Wieben, E.D. Abnormal processing of the human cholecystokinin receptor gene in association with gallstones and obesity. Gastroenterology 1995, 109, 1375–1380.
- Wang, D.Q.; Schmitz, F.; Kopin, A.S.; Carey, M.C. Targeted disruption of the murine cholecystokinin-1 receptor promotes intestinal cholesterol absorption and susceptibility to cholesterol cholelithiasis. J. Clin. Invest. 2004, 114, 521–528.
- Srivastava, K.; Srivastava, A.; Kumar, A.; Mittal, B. Significant association between toll-like receptor gene polymorphisms and gallbladder cancer. Liver Int. 2010, 30, 1067–1072.
- Vishnoi, M.; Pandey, S.N.; Choudhuri, G.; Mittal, B. IL-1 gene polymorphisms and genetic susceptibility of gallbladder cancer in a north Indian population. Cancer Genet. Cytogenet. 2008, 186, 63–68.
- Hsing, A.W.; Sakoda, L.C.; Rashid, A.; Andreotti, G.; Chen, J.; Wang, B.S.; Shen, M.C.; Chen, B.E.; Rosenberg, P.S.; Zhang, M.; et al. Variants in inflammation genes and the risk of biliary tract cancers and stones: A population-based study in China. Cancer Res. 2008, 68, 6442–6452.
- Srivastava, A.; Pandey, S.N.; Choudhuri, G.; Mittal, B. CCR5 Delta32 polymorphism: Associated with gallbladder cancer susceptibility. Scand. J. Immunol. 2008, 67, 516–522.
- Cha, P.C.; Zembutsu, H.; Takahashi, A.; Kubo, M.; Kamatani, N.; Nakamura, Y. A genome-wide association study identifies SNP in DCC is associated with gallbladder cancer in the Japanese population. J. Hum. Genet. 2012, 57, 235–237.
- Stephen, A.E.; Berger, D.L. Carcinoma in the porcelain gallbladder: A relationship revisited. Surgery 2001, 129, 699–703.
- Morimoto, M.; Matsuo, T.; Mori, N. Management of Porcelain Gallbladder, Its Risk Factors, and Complications: A Review. Diagnostics 2021, 11, 1073.
- Lam, R.; Zakko, A.; Petrov, J.C.; Kumar, P.; Duffy, A.J.; Muniraj, T. Gallbladder Disorders: A Comprehensive Review. Dis. Mon. 2021, 67, 101130.
- Berger, M.; Dy, B.; McKenzie, T.; Thompson, G.; Wermers, R.; Lyden, M. Rates of hypercalcemia and hyperparathyroidism among patients with porcelain gallbladder. Am. J. Surg. 2020, 220, 127–131.