Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Amina Yu and Version 3 by Amina Yu.
Abiraterone is the only (as of 2021) CYP17A1 inhibitor approved for the treatment of castrate resistant prostate cancer. It is a molecule based on a steroidal scaffold.
- cytochrome P450 17A1
- CYP17A1
1. Introduction
Current discovery efforts towards new therapies focus on androgen receptor (AR) signaling. Those endeavors introduced the next-generation AR antagonists represented by enzalutamide and apalutamide [1]. Other notable discoveries include proteolysis-targeting chimeras (PROTACs), poly ADP-ribose polymerase (PARP) [2] inhibitors, histone deacetylase (HDAC) inhibitors [3], and various forms of immunotherapy [4]. The emergence of novel targets such as fatty-acid binding protein 5 (FABP5) [5] also illustrates the progress that has been made in the field of prostate cancer. However, despite these efforts, PCa still presents a significant problem.
In addition to the interventions mentioned above, cytochrome P450 17A1 (CYP17A1) inhibition has in recent years received increased attention as a valid treatment modality. CYP17A1 is a dual-function oxygenase membrane-bound enzyme that catalyzes the biosynthesis of steroids [6][7][8]. Its dual activity stems from the ability to produce precursors for glucocorticoids via 17α-hydroxylase reaction and androgens/estrogens via 17,20-lyase reaction [9]. Therefore, CYP17A1 is an attractive target for the treatment of prostate cancers that proliferate in response to androgens [10]. CYP17A1 is required in both the “classic” and “back-door” pathways of steroid biosynthesis, and by its inhibition, the production of androgens can be limited [11][12].
2. Synthesis
A total of 20 new compounds were synthesized using the previously described method [13] (Schemes 1 - 5).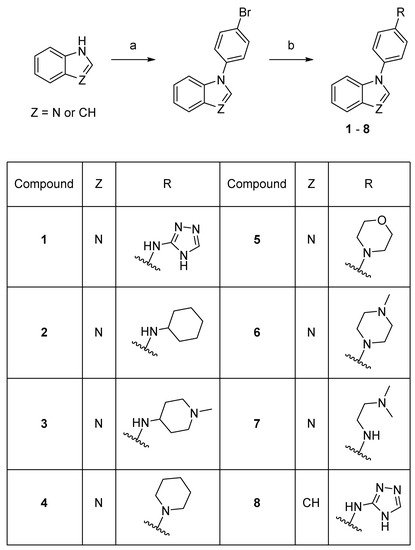
Scheme 1. Synthesis of compounds 1–8. Reaction conditions: (a) 1-bromo-4-fluorobenzene, K3PO4, DMF, 150–160 °C, 53–78%; (b) amine, precatalyst Pd G1 or G3, tBuXPhos, NaOtBu, THF or tBuOH, 21–73%.
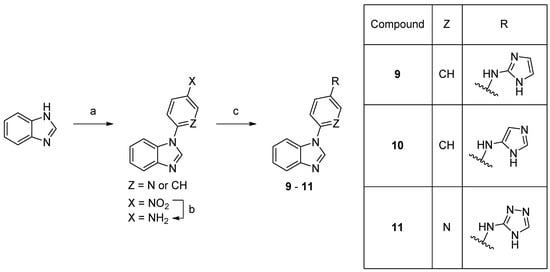
Scheme 2. Synthesis of compounds 9–11. Reaction conditions: (a) 1-fluoro-4-nitrobenzene, K3PO4, DMF 150 °C, 66% or 2-fluoro-5-nitropyridine, K3PO4, DMSO, rt, 74%; (b) 10% Pd/C, MeOH, rt, 82%; (c) bromide, precatalyst Pd G3, tBuBrettPhos, LHMDS, THF, 60 °C, 4–15%.
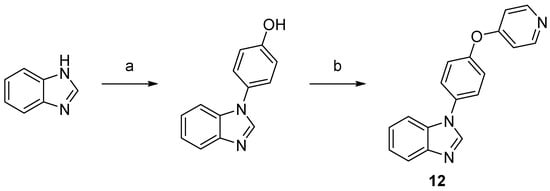
Scheme 3. Synthesis of compound 12. Reaction conditions: (a) 4-hydroxyphenylboronic acid, O2, Cu2S, TMEDA, MeOH, rt, 71%; (b) 4-bromopyridine hydrochloride, NaOtBu, DMF, 150 °C, 32%.
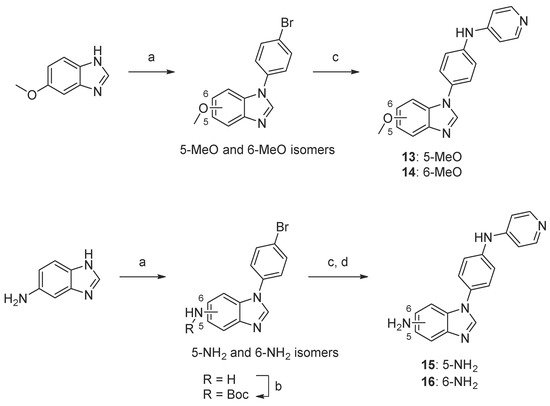
Scheme 4. Synthesis of compounds 13–16. Reaction conditions: (a) 1-bromo-4-fluorobenzene, K3PO4, DMF, 160 °C, 5–44%; (b) Boc2O, TEA, tBuOH, 40 °C, 74–90%; (c) 4-aminopyridine, precatalyst Pd G1, tBuXPhos, NaOtBu, tBuOH, 70 °C, 11–50%; (d) TFA, DCM, 23–50% (over two steps, c and d).
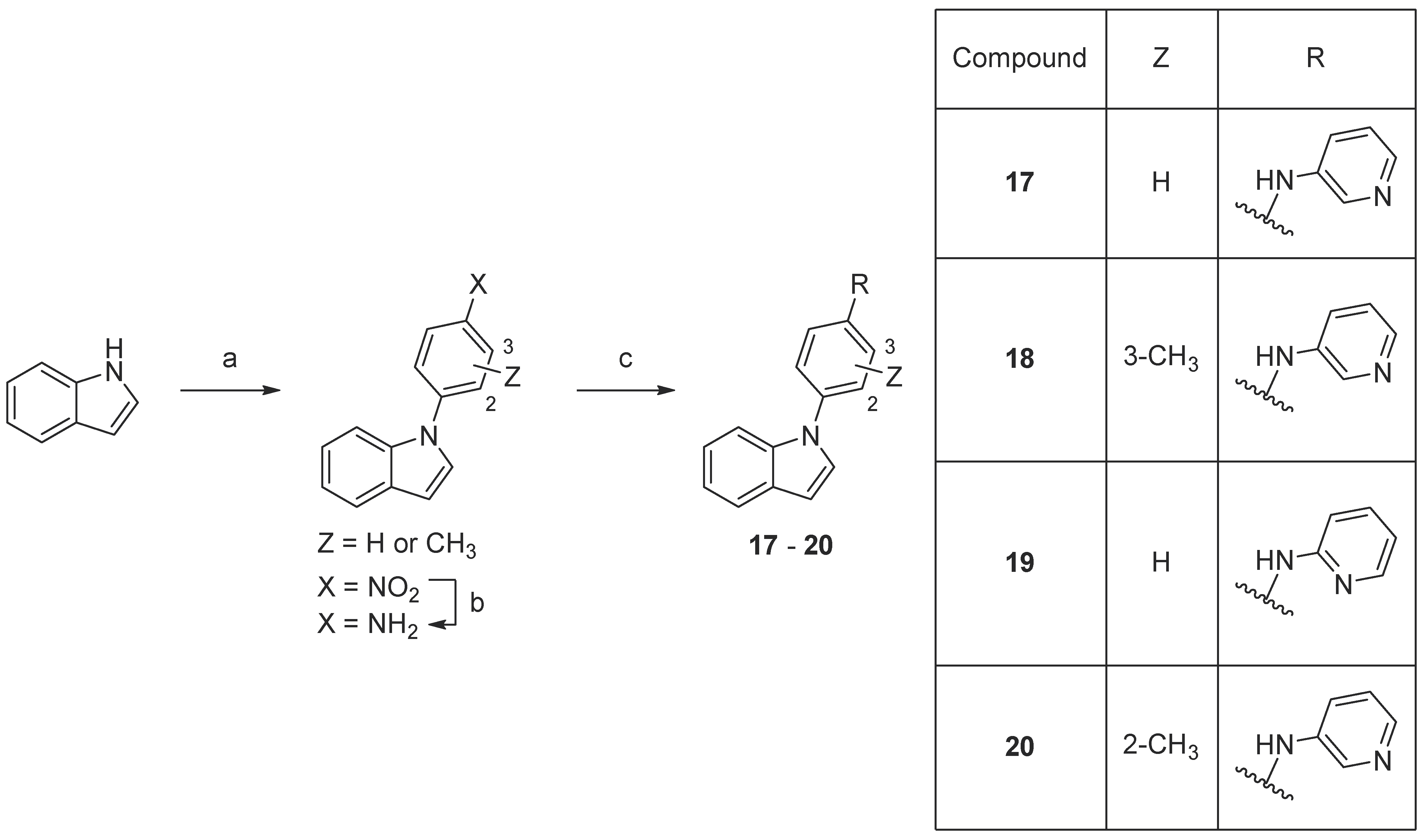
Scheme 5. Synthesis of compounds 17–20. Reaction conditions: (a) 1-fluoro-4-nitrobenzene or fluoronitrotoluene, K3PO4, DMF, 160 °C, 18–93%; (b) 10% Pd/C, MeOH, rt, 91–96%; (c) 2- or 4-aminopyridine, precatalyst Pd G3, tBuXPhos, NaOtBu, THF, MW 100 °C, 23–32%.
The final compounds were fully characterized by 1HNMR and 13CNMR spectroscopy, as well as HRMS spectrometry, and were found to be >99% pure (HPLC).
3. Biological Evaluation
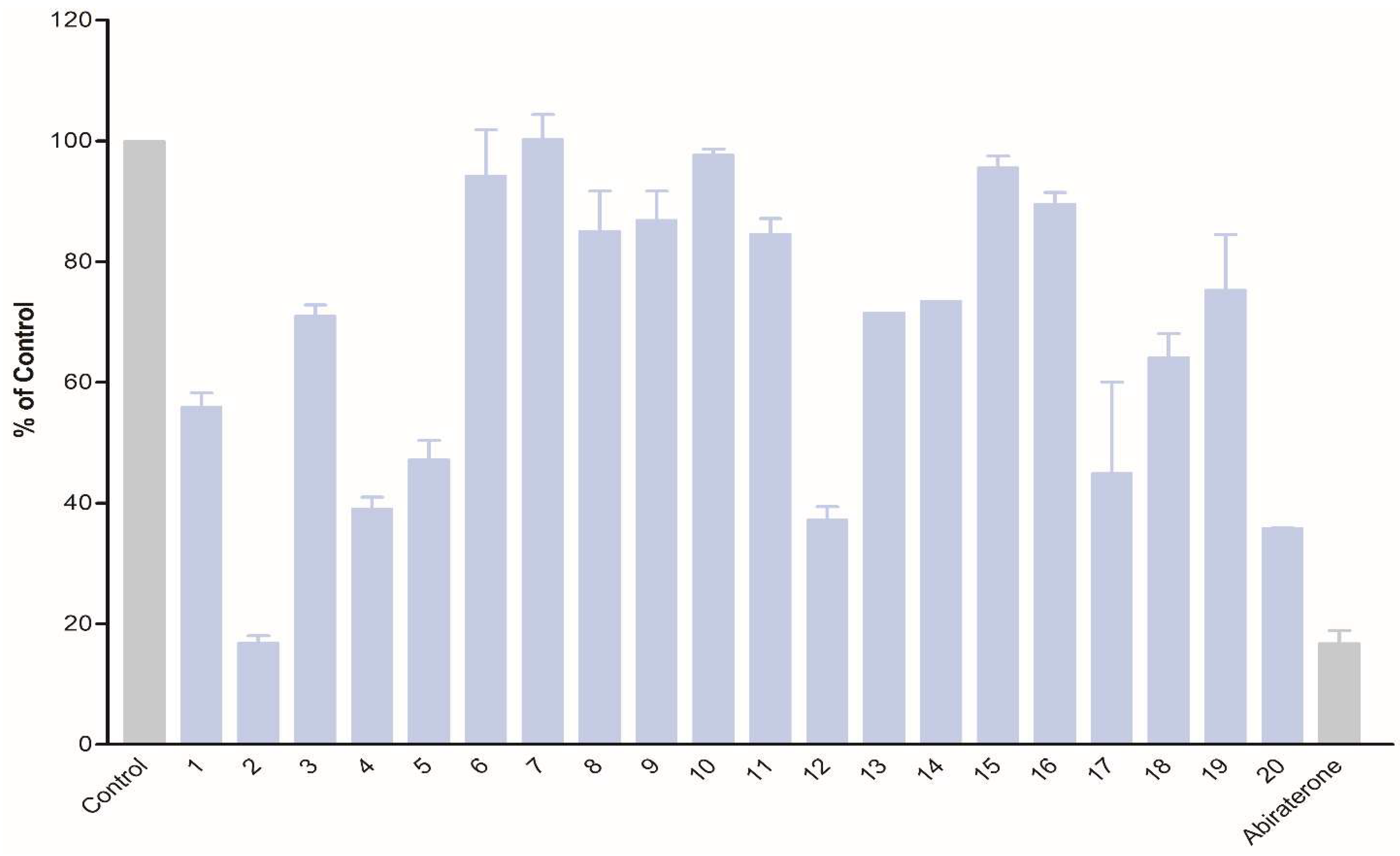
The obtained compounds were subjected to initial screening for inhibition of CYP17A1 activity at a fixed concentration (10 µM) (Figure 1). Substantial activity was observed in the case of compounds 2, 12, and 20. Compound 2 was the most potent and comparable to abiraterone used as a reference compound. These three most potent compounds were selected for more rigorous testing comprising determination of IC50, selectivity towards CYP isoform CYP3A4, the ability to inhibit cytochrome P450 reductase (POR) and the ability to inhibit the lyase reaction of CYP17A1. Compounds 2, 12 and 20 displayed IC50 of 1.2, 3.4 and 2.6 µM, respectively, in the inhibition of the CYP17A1 17α-hydroxylase activity (Figure 2). All three selected compounds exhibited selectivity vs. CYP3A4 (2: 104 ± 4%, 12: 75 ± 5% and 20: 117 ± 8% compared to untreated control) and importantly they do not appear to inhibit POR (2: 93 ± 9%, 12: 103 ± 9% and 20: 88 ± 16% compared to untreated control) (Figure 3), suggesting a targeted activity towards CYP17A1. Finally, checking compounds 2, 12 and 20 against the lyase reaction, unfortunately, revealed little influence compared to abiraterone (2: 78 ± 10%, 12: 77 ± 4% and 20: 82 ± 5% of untreated control, compared to 5 ± 2% activity observed for abiraterone) (Figure 4).
Figure 1.
Inhibition of CYP17A1 17α-hydroxylase activity by compounds
1
–
20
at 10 µM concentration.

Figure 2. Determination of IC50 of compounds 2, 12, and 20 for inhibition of the 17α-hydroxylase activity of CYP17A1.
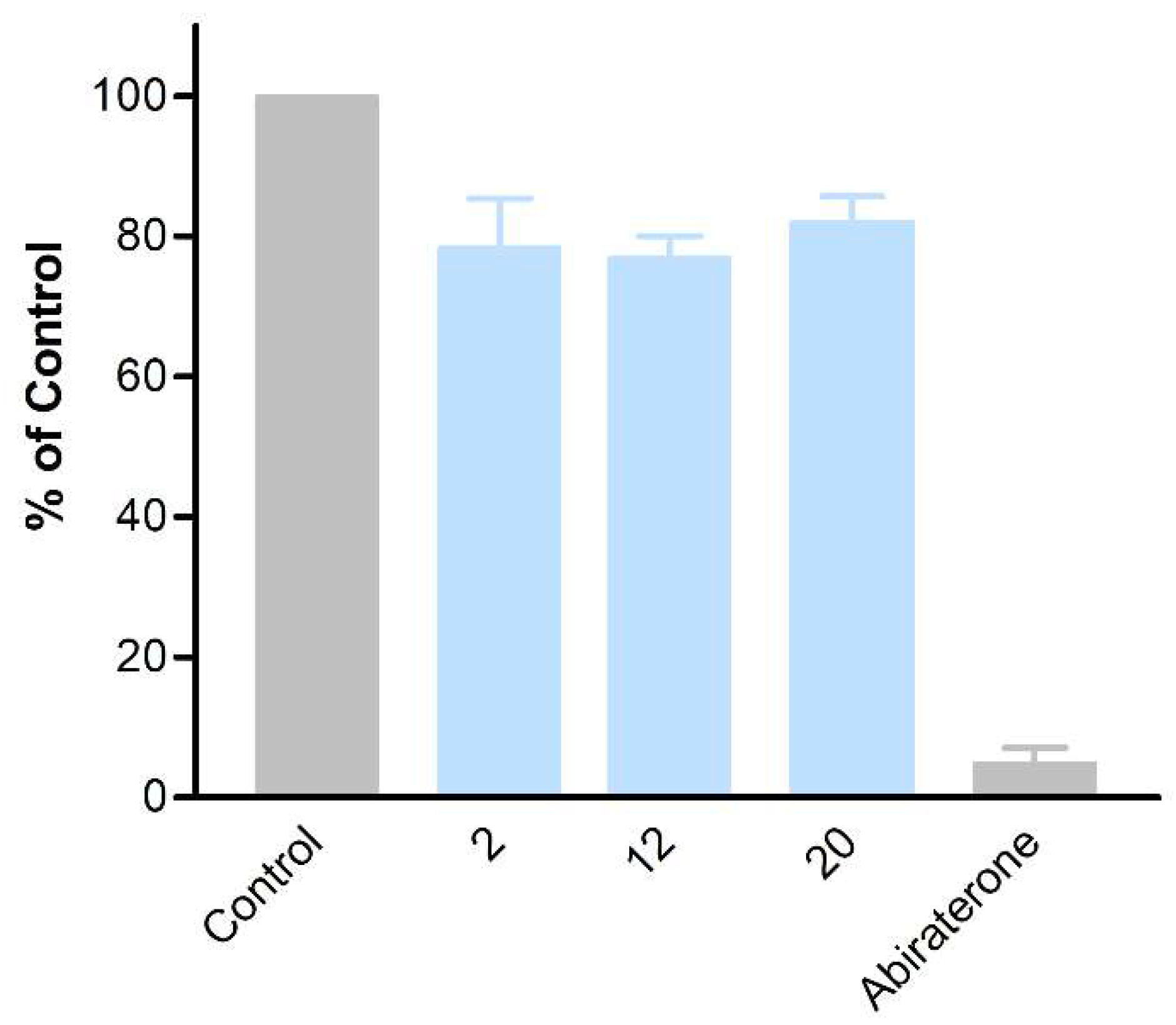

Figure 4. The activity of compounds 2, 12, and 20 towards POR (A) and CYP3A4 (B).
Figure 5. Inhibition of CYP17A1 lyase reaction by compounds 2, 12, and 20.
4. Molecular Modeling
The three compounds, 2, 12, and 20, showing the highest affinity for the CYP17A1 enzyme were subjected to a molecular modeling analysis to determine their potential binding mode. The compounds represent different combinations of ring systems yielding three different scaffolds. Compounds 2 and 20 only contain a single potential heme-coordinating moiety, a benzimidazole and a pyridine moiety, respectively. Compound 12 contains both these ring systems and, accordingly, this compound may bind to CYP17A1 in two different modes. The docking of 2 yields poses with the nitrogen lone pair on the benzimidazole ring pointing towards the Fe atom in the heme moiety (Fe···N = 2.5 Å). The binding mode of 2 is similar to the binding mode it was previously identified for the structurally related compound (1d in [13]) with the cyclohexane ring located in a shallow hydrophobic cavity formed by Leu105, Ile205, and Ile206, which confirms that the enzyme may accommodate different hydrophobic moieties in this part of the active site (Figure 6A). The benzimidazole ring is also located nearly identical to the benzimidazole ring in the CYP17A1–galeterone complex (PDB 3SWZ) [6]. It also observed hydrogen bonding between Asp298 and the amine situated between the aromatic and aliphatic rings (O···N = 3.1 Å). Hydrogen bonding to Asp298 has also been observed in the structure of (S)-orteronel [14] complexed with CYP17A1 (PDB 5IRQ) [15]. Recently, Fehl et al. designed inhibitors derived from abiraterone with polar substituents on the B ring in the steroid framework, which formed hydrogen bonds to Asp298 (PDB 6CHI and 6CIR) [16].
Figure 6. Preferred binding mode predicted by the GOLD(Genetic Optimization for Ligand Docking) docking program of compound 2 (green) (A), 20 (green) in comparison with abiraterone (cyan) (B) and 12 in two docking poses (pose #1 green and pose #2 cyan) (C). Coloring: protein is beige; heme is yellow; and all heteroatoms are colored based on type.
The docking of 20 showed, as expected, that the pyridine nitrogen coordinated to the Fe atom in the heme group with a Fe···N distance of 2.5 Å (Figure 6B). Comparison with the X-ray structures of the CYP17A1–abiraterone complexes (PDB 3RUK and 4NKV) [6][17] revealed that the pyridine rings in the two structures overlap and that the benzene ring in 20 occupies the same space as the C ring of the steroid moiety of abiraterone. The methyl group in 20 was originally introduced to fill the hydrophobic cavity occupied by the B ring of abiraterone. It is possible that the position of the indole moiety relative to the benzene ring is caused by steric repulsion from that methyl group. Another possibility relies simply on a better fit to the upper part of the cavity. In any case, the indole moiety occupies the same space as the A ring and the C10 methyl group in the abiraterone structure. The docking of 20 also suggests that adding a polar substituent on the indole ring could make hydrogen bonding to Asn202, as observed in the CYP17A1–abiraterone complexes.
Compound 12 contains both a pyridine and a benzimidazole system and, accordingly, two possible binding modes are theoretically possible (Figure 6C). The docking revealed these with moieties coordinating to the Fe in the heme group with Fe···N = 2.4 Å (benzimidazole) and 2.5 Å (pyridine), respectively, referred to as pose #1 and pose #2. For compound 12, pose #1 the benzimidazole system is located similarly to the benzimidazole system in the galeterone complex (3SWZ) [6], but in the opposite end of the compound, the pyridine is not close enough to make a contact to Asn202 as observed for galeterone. There are distinct differences in the orientation of the pyridine ring in pose #2 and in the abiraterone complexes 3RUK and 4NVK relative to the heme system. The 4-substituted pyridine ring requires more space above the heme group to adopt an ideal orthogonal orientation [18]. It suggests that the 3-substituted pyridine is the preferred moiety relative to the 2- and 4-substituted systems.
References
- Velho, P.I.; Anna, P.T.S.; da Silva, R.F.P.; Ferreira, R.D.P.; Venero, F.C. The development of apalutamide for the treatment of prostate cancer. Expert Opin. Drug Discov. 2021, 16, 217–226.
- Risdon, E.N.; Chau, C.H.; Price, D.K.; Sartor, O.; Figg, W.D. PARP Inhibitors and Prostate Cancer: To Infinity and Beyond BRCA. Oncologist 2021, 26, e115–e129.
- Rana, Z.; Diermeier, S.; Hanif, M.; Rosengren, R.J. Understanding Failure and Improving Treatment Using HDAC Inhibitors for Prostate Cancer. Biomedicines 2020, 8, 22.
- Rathi, N.; McFarland, T.R.; Nussenzveig, R.; Agarwal, N.; Swami, U. Evolving Role of Immunotherapy in Metastatic Castration Refractory Prostate Cancer. Drugs 2021, 81, 191–206.
- O’Sullivan, S.E.; Kaczocha, M. FABP5 as a novel molecular target in prostate cancer. Drug Discov. Today 2020, 25, 2056–2061.
- DeVore, N.M.; Scott, E.E. Structures of cytochrome P450 17A1 with prostate cancer drugs abiraterone and TOK-001. Nature 2012, 482, 116–119.
- Nakajin, S.; Hall, P.F.; Onoda, M. Testicular microsomal cytochrome P-450 for C21 steroid side chain cleavage. Spectral and binding studies. J. Biol. Chem. 1981, 256, 6134–6139.
- Chung, B.C.; Picado-Leonard, J.; Haniu, M.; Bienkowski, M.; Hall, P.F.; Shively, J.E.; Miller, W.L. Cytochrome P450c17 (steroid 17 alpha-hydroxylase/17,20 lyase): Cloning of human adrenal and testis cDNAs indicates the same gene is expressed in both tissues. Proc. Natl. Acad. Sci. USA 1987, 84, 407–411.
- Nakajin, S.; Shively, J.E.; Yuan, P.M.; Hall, P.F. Microsomal cytochrome P-450 from neonatal pig testis: Two enzymatic activities (17 alpha-hydroxylase and c17,20-lyase) associated with one protein. Biochemistry 1981, 20, 4037–4042.
- Yin, L.; Hu, Q. CYP17 inhibitors—abiraterone, C17,20-lyase inhibitors and multi-targeting agents. Nat. Rev. Urol. 2013, 11, 32–42.
- Bird, I.M.; Abbott, D.H. The hunt for a selective 17,20 lyase inhibitor; learning lessons from nature. J. Steroid Biochem. Mol. Biol. 2016, 163, 136–146.
- Flück, C.E.; Meyer-Boni, M.; Pandey, A.V.; Kempna, P.; Miller, W.L.; Schoenle, E.J.; Biason-Lauber, A. Why boys will be boys: Two pathways of fetal testicular androgen biosynthesis are needed for male sexual differentiation. Am. J. Hum. Genet. 2011, 89, 201–218.
- Wróbel, T.M.; Rogova, O.; Andersen, K.L.; Yadav, R.; Brixius-Anderko, S.; Scott, E.E.; Olsen, L.; Jørgensen, F.S.; Björkling, F. Discovery of Novel Non-Steroidal Cytochrome P450 17A1 Inhibitors as Potential Prostate Cancer Agents. Int. J. Mol. Sci. 2020, 21, 4868.
- Yamaoka, M.; Hara, T.; Hitaka, T.; Kaku, T.; Takeuchi, T.; Takahashi, J.; Asahi, S.; Miki, H.; Tasaka, A.; Kusaka, M. Orteronel (TAK-700), a novel non-steroidal 17,20-lyase inhibitor: Effects on steroid synthesis in human and monkey adrenal cells and serum steroid levels in cynomolgus monkeys. J. Steroid Biochem. Mol. Biol. 2012, 129, 115–128.
- Petrunak, E.M.; Rogers, S.A.; Aubé, J.; Scott, E.E. Structural and Functional Evaluation of Clinically Relevant Inhibitors of Steroidogenic Cytochrome P450 17A1. Drug Metab. Dispos. 2017, 45, 635–645.
- Fehl, C.; Vogt, C.D.; Yadav, R.; Li, K.; Scott, E.E.; Aubé, J. Structure-Based Design of Inhibitors with Improved Selectivity for Steroidogenic Cytochrome P450 17A1 over Cytochrome P450 21A2. J. Med. Chem. 2018, 61, 4946–4960.
- Petrunak, E.M.; DeVore, N.M.; Porubsky, P.R.; Scott, E.E. Structures of Human Steroidogenic Cytochrome P450 17A1 with Substrates. J. Biol. Chem. 2014, 289, 32952–32964.
- Bonomo, S.; Hansen, C.H.; Petrunak, E.M.; Scott, E.E.; Styrishave, B.; Jørgensen, F.S.; Olsen, L. Promising Tools in Prostate Cancer Research: Selective Non-Steroidal Cytochrome P450 17A1 Inhibitors. Sci. Rep. 2016, 6, 29468.
More