Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Carrie Welch and Version 4 by Jason Zhu.
Pulmonary arterial hypertension (PAH) is a rare, progressive vasculopathy with significant cardiopulmonary morbidity and mortality. Roles for rare variants in three channelopathy genes—ABCC8, ATP13A3, and KCNK3—have been validated in multiple PAH cohorts, and in aggregate explain ~2.7% of PAH cases. Complete or partial loss of function has been demonstrated for PAH-associated variants in ABCC8 ABCC8 and KCNK3. Channels can be excellent targets for drugs, and knowledge of mechanisms for channel mutations may provide an opportunity for the development of PAH biomarkers and novel therapeutics for patients with hereditary PAH but also potentially more broadly for all patients with PAH.
- channelopathy
- genetics
- lung disease
- pulmonary arterial hypertension
1. ATP-Binding Cassette Subfamily Member 8 (ABCC8)
Outside of the TGFβ/BMP pathway, channelopathy gene, ABCC8, is one of the most common causes of pulmonary arterial hypertension (PAH), accounting for ~1% of cases. ABCC8 is a member of the ATP-binding cassette (ABC) transporter gene superfamily and encodes the sulfonylurea receptor 1 (SUR1) protein, an ATP-sensitive potassium channel regulatory subunit. Potassium channels play important roles in maintaining cellular resting membrane potential and intracellular calcium concentrations. ABCC8 is highly expressed in pancreatic islet cells where it functions to release insulin, and recessive mutations cause congenital hyperinsulinemia and neonatal diabetes mellitus [1][2][23,24]. ABCC8 was first identified as a PAH risk gene by exome sequence analysis of a cohort of 99 pediatric- and 134 adult-onset PAH cases [3][10]. A rare, predicted deleterious de novo missense variant was identified in a 10-year-old patient with IPAH. De novo variants are infrequent and, when deleterious, can be associated with high mortality at an early age and, therefore, are not transmitted to the next generation. Thus, the entire PAH cohort was screened for genetic variants in ABCC8, and rare or novel missense variants were identified in seven unrelated patients with IPAH, HPAH or APAH-congenital heart disease (APAH-CHD). Consistent with autosomal dominant inheritance of PAH, all individuals were heterozygous for the rare ABCC8 variants. The ABCC8 c.718G>A;p.240A>T variant segregated with PAH in one family. A replication study in a United Kingdom (UK) PAH cohort identified four additional variants, three missense and one splice variant. One ABCC8 variant heterozygote also had a rare TBX4 frameshift mutation; and none of the other carriers had mutations in any known risk genes. In the combined US/UK, approximately half of the patients had adult-onset and half pediatric-onset disease. Statistical enrichment analyses of ABCC8 variants in European PAH cases compared to two independent European population control groups (n > 33,000 individuals each), indicated a 3-fold enrichment rate of ABCC8 variants among PAH cases with no significant difference in the frequency of predicted benign synonymous variants between cases and controls.
Independent studies carried out in large PAH cohorts from the US and Spain and two smaller cohorts have validated ABCC8. The US-based National Biological Sample and Data Repository for PAH (aka PAH Biobank) includes 2572 PAH cases with exome sequence data [4][6]. Rare or novel, predicted deleterious ABCC8 missense variants were identified in twenty-eight cases. There were twenty-five unique variants, with three recurrent variants (Met400Val, Arg670Cys, Gln830Lys) each identified in two unrelated cases. Additionally, novel missense variant, c.2439T>A;p.Ser813Arg, affects the same amino acid residue as c.2437G>A;p.Ser813Arg reported in Bohnen et al. [3][10]. The ABCC8 variants occurred at the same frequency in IPAH and APAH (CHD, connective tissue diseases, and one case with HIV). Ten percent (n = 3) of the cases were diagnosed as children, similar in frequency to the overall composition of the cohort (91% adult, 9% children). Panel gene sequencing of 21 PAH genes in 624 PAH cases from the National Spanish PAH Registry identified five missense, one splice, and one frameshift variant were identified in ABCC8 [5][6] [11,25]. The splice variant and one missense variant predicted to affect splicing were demonstrated to cause exon skipping in a minigene assay [6][25]. The splice variants were identified in patients with APAH (CREST, CHD); the frameshift variant carrier had IPAH, and the missense variant heterozygotes had IPAH (n = 4) or APAH-CHD (n = 1). In a small cohort of pediatric PAH cases (n = 18), Gelinas and colleagues identified a novel ABCC8 missense variant associated with IPAH in an analysis of 26 PAH genes [7][12]. Finally, in a Chinese cohort of persistent pulmonary hypertension of the newborn (n = 74), Liu and colleagues identified a novel stopgain variant, c.2331G>A;p.W777X, with exome sequencing in a newborn male with hypoglycemia [8][13]. Overall, ABCC8 variants explain ~1.4% of PAH cases, including primary PAH and PAH associated with other diseases.
Of the 45 rare, predicted deleterious ABCC8 variants identified in 49 unrelated PAH cases, 41 are missense variants. The locations of the missense variants, one frameshift, and one stopgain variant are shown in Figure 1. Variants identified in adult- and pediatric-onset PAH are depicted above and below the protein schematic, respectively. The vast majority of the missense variants (32/41) reside in conserved ABC protein transmembrane or nucleotide-binding domains. Three additional variants, c.647G>A;p.Arg216His, c.686C>T;p.Thr229Ile and c.718G>A;p.Ala240Thr, reside in a cytoplasmic loop containing a Lasso motif near the ATP site which may regulate channel activity [9][26]. Eight of the missense variants have been tested via patch-clamp electrophysiology and rubidium efflux studies of mutant COS cells, and all mutant cells exhibited significantly decreased channel activity [3][10]. Diazoxide is a SUR1 activator used to treat congenital hyperinsulinemia. Addition of exogenous diazoxide to the ABCC8 mutant cells at least partially normalized currents and rubidium efflux [3][10]. The single-frameshift and stopgain variants are predicted to result in nonsense-mediated decay. Some of the PAH-associated ABCC8 variants have been reported in patients with autosomal recessive juvenile hyperinsulinemia or neonatal diabetes mellitus but none of the heterozygous PAH cases, aside from the case with persistent pulmonary hypertension of the newborn, exhibited derangements in glucose metabolism. Diabetes is a common metabolic comorbidity associated with adult-onset PAH [10][27]; whether heterozygous variants in ABCC8 contribute to the co-occurrence of diabetes and adult-onset PAH remains an open question.
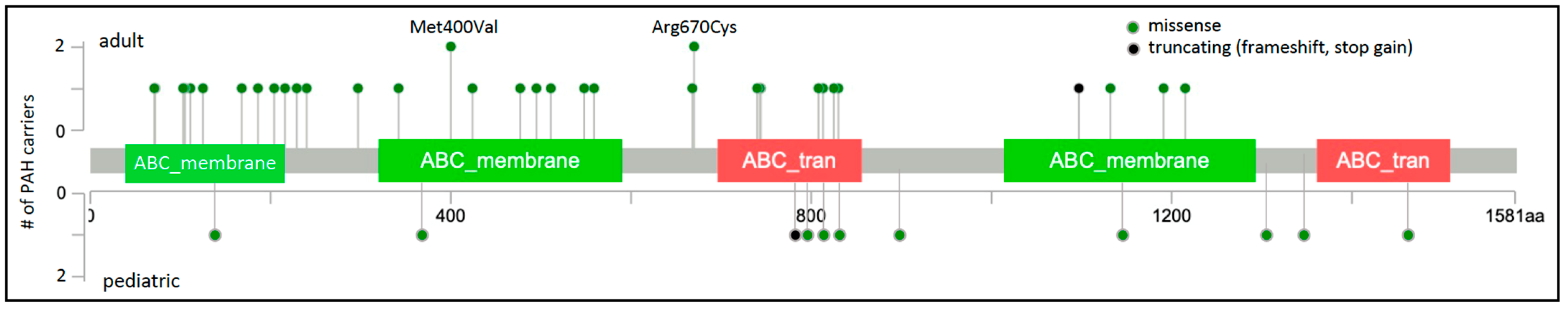
Figure 1. ABCC8 two-dimensional protein schematic. Variants identified in adult-onset PAH cases are shown above the protein schematic; variants identified in children are shown below. Variant type is color-coded. The number of PAH carriers identified with a particular variant is shown along the y-axis. Note that splice variants are not included and variant density may impede visualization of closely-located variants. Conserved protein domains are indicated by colored rectangles. ABC_membrane, ABC transporter transmembrane region; ABC_tran, ABC transporter.
2. ATPase 13A3 (ATP13A3)
ATP13A3 encodes a transmembrane cation transporter which was recently shown to transport polyamines [11][31]. Polyamines are small metabolites required for normal cell growth and proliferation, and elevated plasma concentrations have been reported in multiple cancers and, more recently, PAH [12][13][32,33]. Genetic variants in ATP13A3 were first reported in a genome sequencing study of the UK NIHR Bioresource-Rare Diseases Study of 1083 PAH cases [14][8]. After excluding cases with variants in seven established PAH causal genes, a higher frequency of protein-truncating variants was observed in cases compared to controls. A trend was also observed for protein-truncating plus missense variants but did not reach genome-wide significance. Ten unrelated IPAH cases with unique rare, predicted deleterious variants were identified: three frameshift, two stopgain, one splice variant, and four missense variants. Independent validation of ATP13A3 in PAH was demonstrated in several studies worldwide. Four additional missense variants were identified in a Chinese cohort of 331 unrelated IPAH cases [15][14]. In the European pediatric PAH cohort cited above, one novel missense variant was identified in an APAH-CHD case [7][12]. Interferon beta is recognized as a cause of drug-induced PAH, often reversible with cessation of treatment [16][34]. In a study of two multiple sclerosis patients treated with IFNβ-1a and exhibiting reversible PAH [17][15], Lerche and colleagues identified a novel missense variant, c.1540C>T;p.Gln514X, in one of the patients. In the PAH Biobank, seven unique variants (three frameshift, one stopgain, and three missense) were identified in seven unrelated cases (five HPAH/IPAH, one APAH-CTD, and one APAH-CHD) [4][6]. In each of these studies, the cases were adult-onset disease, with the exception of one child, and all were heterozygous.
It has beWen reported a case series of three families with pediatric-onset severe PAH and early mortality [18][16]. In total, there were five children diagnosed with PAH under the age of three years, largely refractory to medical treatment, and four died in early childhood. The surviving child underwent a Pott’s shunt to decompress the right ventricle [19][35]. Exome/genome sequence identified biallelic ATP13A3 variants in each of the affected children. The unaffected parents were heterozygous for one of the variants, and unaffected siblings were either heterozygous for a variant or homozygous for the reference allele. The variants included two frameshift, one stopgain, and two missense variants. Variants occurring at ATP13A3 ATP13A3 amino acid 850 have been reported now in three cases, including three occurrences of frameshift variant c.2549dup; p.Met850Ilefs*13 (a monoallelic adult case [4][6] and a biallelic childhood case [18][16]). Further, c.2228G>T;p.Arg743Cys, identified in a biallelic child-onset case, impacts the same amino acid residue as reported for an adult-onset case (c.2227C>T;p.Arg743Cys) [4][6]. These data are consistent with semi-dominant inheritance for the ATP13A3 gene. Biallelic inheritance indicates a dose-dependent effect and may have implications for prognosis and aggressive treatment strategies in child-onset PAH.
The protein locations of the 23 unique ATP13A3 variants are shown in Figure 2. ATP13A3 is highly constrained for loss-of-function variants (pLoF = 1) [20][36], with heterozygous alleles occurring very rarely and no homozygous occurrences reported in gnomAD. Most of the missense variants (7/10) occur in conserved protein domains. A role for ATP13A3 in polyamine transport has only recently been reported [11][31], and is currently under investigation. ATP13A3 is widely expressed in the developing embryo and adult tissues [21][37], including PASMCs [14][8]. Hypoxia stimulates accumulation of spermine leading to increased PASMC proliferation in model systems [33]. We [13]hypothesize that ATP13A3 variants predicted to alter transporter function disturb polyamine homeostasis.
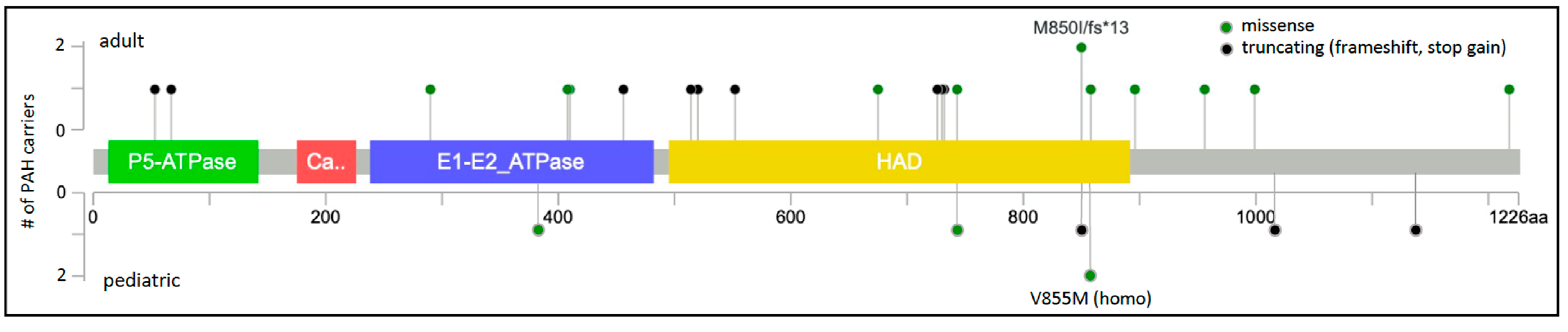
Figure 2. ATP13A3 two-dimensional protein schematic. Variants identified in adult-onset PAH cases are shown above the protein schematic; variants identified in children are shown below. Variant type is color-coded. The number of PAH carriers identified with a particular variant is shown along the y-axis. Conserved protein domains are indicated by colored rectangles. E1-E2_ATPase, cation transporter ATPase; HAD, haloacid dehalogenase-like hydrolase.
3. Potassium Two-Pore Domain Channel (KCNK3)
The KCNK3 gene encodes a two-pore domain potassium channel, also known as TASK1. KCNK3 was first identified as a PAH causal gene by rare variant analysis of exome sequencing data in a family with five affected members [22][17]. A c.608G>A;p.Glu203Asp missense variant was identified in four affected and one unaffected family members. An additional unaffected individual did not carry the family variant. To identify additional KCNK3 variants, exome sequencing data from 92 familial and 230 IPAH cases were screened. Five additional heterozygous missense variants were identified, confirming an autosomal dominant mode of inheritance with incomplete penetrance. None of these KCNK3 variant heterozygotes had variants in other known PAH risk genes. Patch clamp studies in COS-7 cells expressing the mutant proteins indicated loss of potassium channel function that could be partially rescued by treatment with a phospholipase inhibitor [22][17]. Similarly decreased channel current resulted from expression of mutant channels alone or co-expression of both mutant and wild-type channels.
Rare missense variants in KCNK3 were subsequently reported in at least six additional studies of HPAH and IPAH. Two novel variants were identified in a Spanish cohort of 136 unrelated cases [23][18]. One of the cases, a severe form of PAH diagnosed at 2 months of age, was from a consanguineous family and was homozygous for the c.316G>C;p.Gly106Arg variant His-affected mother was heterozygous for the variant and diagnosed at 19 years of age. Functional analysis of the variants showed similar membrane localization and loss of channel function that could not be rescued by pharmacologic activators of TASK-1 channels [24][38]. An exome/genome sequencing analysis of nine Japanese families with PAH identified one rare missense variant in one family [25][19], c.608G>A;p.Glu203Asp, a recurrence of the variant first observed in a family from Ma et al. [22][17]. Screening of 82 Chinese pediatric PAH cases [26][20] revealed another recurrent KCNK3 variant, c.289G>A;p.Gly97Arg, also reported by Ma et al. [22][17]. Exome sequence analysis of 412 largely European pediatric and adult cases with HPAH or IPAH identified one case with a novel variant, c.675C>A;p.Phe225Ile, and one case with recurrent variant, c.544G>A;p.Glu182Lys [27][21]. A single occurrence of the c.544G>A;p.Glu182Lys variant was also identified in the PAH Biobank [4][6]. Finally, a different coding variant affecting the same amino acid residue, c. 544G>A;p.Glu182Gln, was identified in a large rare disease cohort from the UK [28][22]. A total of nine variants in fourteen cases, with four recurrent variants identified in at least two cases each, were identified in these seven studies. Together, the variants, all missense, explain ~0.3% of HPAH/IPAH (Table 1). The locations of the nine PAH-associated KCNK3 variants are depicted in Figure 3; all but one variant maps to one of two conserved ion transport domains. The high frequency of recurrence of PAH-associated KCNK3 variants is striking, underscoring the importance of these residues in pulmonary vascular dysfunction. While a semi-dominant mode of inheritance is indicated in the consanguineous family with very early-onset severe PAH, segregation data in all other families indicate an autosomal dominant mode.
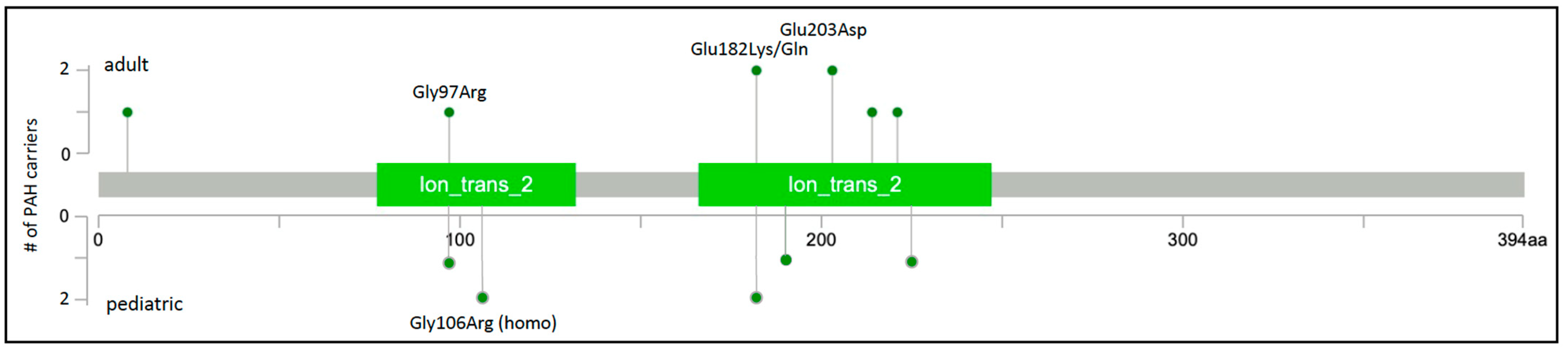
Figure 3. KCNK3 two-dimensional protein schematic. Variants identified in adult-onset PAH cases are shown above the protein schematic; variants identified in children are shown below. All variants are missense. The number of PAH carriers identified with a particular variant is shown along the y-axis. Conserved protein domains are indicated by colored rectangles. Ion_trans_2, ion channel.