Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Yassein Mozamel Ibrahim and Version 3 by Amina Yu.
Porcine sapelovirus (PSV) is an important emerging pathogen associated with a wide variety of diseases in swine, including acute diarrhoea, respiratory distress, skin lesions, severe neurological disorders, and reproductive failure.
- porcine sapelovirus
- isolation
- characterization
- prevalence
- ELISA
- monoclonal antibodies
1. Introduction
Sapelovirus is a new genus within the family Picornaviridae. Currently, this genus comprises porcine Sapelovirus (PSV), simian Sapelovirus, avian Sapelovirus, and unclassified Sapelovirus isolated from bat, marmot, California sea lion, and mouse [1]. The Sapelovirus genome is a positive-strand RNA of approximately 7.5 kb in length, with the typical picornavirus genome organization: a 5′ untranslated region (UTR), a single large open reading frame (ORF), a 3′-UTR, and a poly (A) tail. The ORF encodes a single polyprotein that is subsequently cleaved by virus encoding proteases into twelve proteins, including leader protein, four structural proteins (VP1-VP4) and seven nonstructural functional proteins (2A, 2B, 2C, 3A, 3B, 3C, and 3D) [2]. The capsid proteins of Sapelovirus are composed of four structural proteins located at the virion surface and exhibit high sequence variability [1]. VP1 protein, the most dominant and variable viral protein, has proven useful in determining genetic relationships among picornaviruses [3][4][5][6][3,4,5,6].
PSV is transmitted through the fecal-oral route and associated with various symptoms, ranging from asymptomatic to clinical diseases such as respiratory distress, acute diarrhea, skin lesions, severe neurological disorders, and reproductive failure in domestic swine and wild boar [7][8][9][10][11][7,8,9,10,11]. Since first reported in UK [12], PSV has been identified in different countries worldwide, with prevalence ranging between 7.1% in India and 71.0% in Hungary [2][13][14][15][16][17][18][19][20][21][2,13,14,15,16,17,18,19,20,21]. Recent reports have shown that PSV can induce intestinal lesions in experimentally infected piglets [1][8][22][1,8,22]. Previous studies have recorded lethal PSV infections in pigs aged 3 to 12 weeks with neurological problems, diarrhea, and respiratory ailments [7][10][11][7,10,11]. The coinfection of viral diarrhea agents complicates disease detection, control, and prevention. Coinfection of PSV with other enteric pathogens is frequently reported in both symptomatic and asymptomatic pigs [4][10][18][20][22][23][24][25][4,10,18,20,22,23,24,25]. Further, the asymptomatic nature of PSV infections and the high coinfection rate cause the symptoms of PSV infection to go unnoticed [24], which poses a considerable risk to swine industries. Because antiviral drug and vaccine are not available yet, early identification and accurate diagnosis play a decisive role in timely containment and control of PSV infection.
Currently, diagnosis of PSV is mainly based on the detection of nucleic acid by PCR and confirmed by virus isolation [1][15][19][1,15,19]. Although detection of PSV by PCR is sensitive, serological diagnosis should be more accurate. However, no specific antibody-based reagent or serological assay is commercially available yet to detect PSV infections. Based on these facts, highly sensitive and effective diagnostic assays are crucial to investigate the epidemiology of PSV.
2. Isolation of PSV
PK15 cells were inoculated with six diarrheic stool samples that were positive to PSV but negative for other enteric viruses, including TGEV, PEDV, PDCoV, RVA, PSaV, PKV, and EVG. After 3 blind passages, cells inoculated with four samples started to show CPEs, characterized by cell rounding, shrinking, and detachment (Figure 1A). After three rounds of plaque purification, the purified virus developed uniform and clear plaques (Figure 1B). After ultracentrifugation, the plaque-purified viruses were examined under TEM, and the results showed that spherical, non-enveloped virus particles of approximately 32 nm in diameter were observed (Figure 1C). The RT-PCR and sequencing results showed that the isolated viruses were PSV. The virus growth curve results showed that the virus replicated fast and reached a peak of 107.8 TCID50/mL at 24 h, suggesting that the cycle of PSV multiplication is completed within 24 h (Figure 1D).
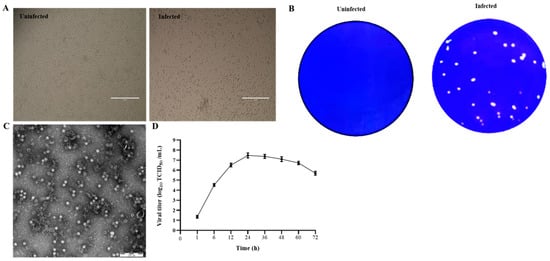
Figure 1. Isolation and identification of PSV. (A) Cytopathic effects in PSV-infected PK15 cells at 24 hpi. (B) Production of plaques of the PSV isolate in PK15 cells. (C) Picornavirus-like particles under TEM. (D) Growth kinetic of PSV.
3. Whole-Genome Sequence Analysis
Using next-generation sequencing technology, four entire genome sequences of PSV were obtained, named as PSV-12, PSV-14, PSV-15, and PSV-41 (GenBank access No. OM037653-OM37656). The results revealed that the entire genome of isolated PSVs ranged from 7569–7572 nucleotides, including 490 nucleotides of 5′-UTR sequence and 83 nucleotides of 3′-UTR sequence. By comparing the genomes of previously identified PSV strains with these four PSV isolates, the full ORF and cleavage sites of PSV were predicted. The length of the ORF of PSV-12, PSV-14, and PSV-15 strains was 6996 nucleotides, encoding 2332 amino acids, whereas, PSV-41 has longer ORF of 6999 nucleotides that encode 2333 amino acids due to a single amino acid insertion at position (896 P) at the 3′-end of ORF.
The nucleotide and amino acid sequence identities among four PSV isolates ranged from 92–99.9% and 98.6–100%, respectively. While PSV-12, PSV-14, and PSV-15 were isolated from the same farm, their nucleotide (99.7–99.8%) and amino acid sequences (99.9–100%) similarities suggest that similar PSV strains were circulating on the farm. In contrast, PSV-41, isolated from another farm, showed a relatively low identity in nucleotide (91.1–91.2%) and amino acid sequences (98.6–98.7%) with the other three PSVs. These data indicate that PSVs circulating on two farms are genetically distinct. Furthermore, identity comparisons of whole genome among PSVs demonstrated that PSVs identified in hthis study had 77.7–92% and 84.3–98.9% similarities in nucleotide and amino acid sequences, respectively, compared to the 66 PSV reference strains available in the GenBank database.
The VP1 gene of three PSV strains (12, 14, and 15) contained 882 nucleotides and encoded 294 (aa) protein, according to an alignment analysis of capsid VP1 of PSVs identified in this study with those of other known PSVs. The PSV-41 strain, on the other hand, had three more nucleotides (885), one extra proline inserted between aa 286 and 287 of the VP1 (Figure 2A). Homology comparison of the VP1 gene of isolated PSVs with reference strains revealed sequence similarity ranging from 62.6% to 90.6% nucleotide and 60.6% to 99% amino acid. Moreover, the isolated PSV strains had a sequence similarity of 90.7–100% nucleotide and 96.4–100% amino acid with each other.

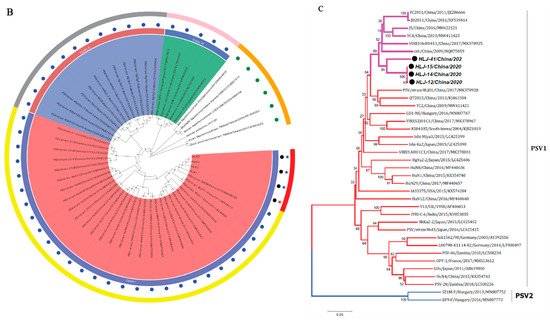
Figure 2. Sequence analysis of the isolated PSV strains. (A) Alignment of 3′-partial amino acid sequences of VP1 of PSV strains. Sequences in boxes showed the hypervariable region in the C-terminus of VP1. The strains identified in the present study were indicated by black dots; (-), missing amino acids. (B,C) Phylogenetic analysis of PSV based on the complete nucleotide sequence and VP1 gene respectively. The trees were generated by using MEGA v.6.0 with the neighbor-joining method with the Kimura 2-parameter with 1000 bootstrap replication. The black circle indicates PSV isolated in this study. The scale bars represent the number of substitutions per site.
4. Phylogenetic Analysis
The phylogenetic tree based on complete ORF nucleotide sequences was constructed using the current PSVs sequences and representative PSV sequences from the NCBI database (Table S2). As shown in Figure 2B, PSVs were phylogenetically classified into three clades (clade I—clade III), correlated with their geographical location. Clade I included Chinese and Zambian PSV strains. Clade II consisted of PSV strains detected in South Korea and Japan, while clade III was composed of the strains identified in French, Germany, India, USA, and UK. The four PSV strains isolated in this study were grouped into clade I and formed a monophyletic clade with Chinese and Zambian strains.
The phylogenetic tree based on VP1 protein revealed that all PSVs were separated into two clades, each corresponding to two genotypes, named PSV-1 and PSV-2 (Figure 2C). The PSV strains isolated here were classified in the typical PSV-1 clade, along with previously described PSV strains; whereas two Hungarian strains, SZ1M-F and EF9-F, were classified in the PSV-2 clade. The four PSV isolates have formed a monophyletic clade, with topological structure and branching of the evolutionary tree in the same manner of complete ORFs.
54. Expression and Purification of VP1 Protein
The VP1 protein of the PSV-14 was expressed as an MBP-tagged fusion protein as shown in the SDS-PAGE result (Figure 3A). The expression of recombinant VP1 protein of approximately 77 kDa was confirmed by Western blot using an MBP-specific antibody (Figure 3B). The recombinant VP1 protein was purified by the AKTA liquid affinity chromatography system using an MBP trap column. The purified recombinant VP1 protein was evaluated by Western blot, and the results showed that the recombinant VP1 protein was efficiently purified (Figure 3C).
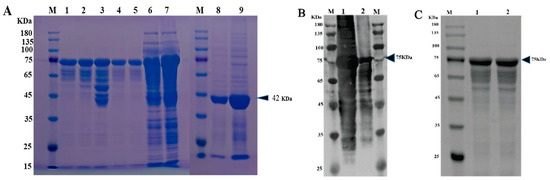
Figure 3. Expression and purification of PSV-VP1 protein in E. coli. (A) SDS-PAGE analysis of the MBP-tagged recombinant VP1-protein expression (~75 KDa). Lane M, protein marker; Lanes 1–5, purified bacterial cell lysates of MBP-tagged VP1 at different concentrations; Lines 6 and 7, unpurified bacterial cell lysate of MBP-tagged VP1; Line 8, sediment of bacterial cell lysate of empty vector, Line 9, supernatant of bacterial cell lysate of =empty vector (~42 KDa). (B) Western blot analysis of unpurified recombinant VP1-protein using anti-MBP tag antibody. Lane 1, 0.2 mg/mL of unpurified VP1 protein; Lane 2, 0.1 mg/mL of unpurified VP1 protein. (C) Western blot analysis of purified VP1-recombinant protein using anti-MBP tag antibody. Lane 1, 0.1 mg/mL of purified VP1 protein; Lane 2, 0.2 mg/mL of purified VP1 protein.
6. Production and Application of mAbs
The mouse with the highest antibody titer against recombinant VP1 was sacrificed, and splenocytes were fused with SP2/0 myeloma cells to create a confluent hybridoma that was screened using the purified recombinant VP1 protein as coating antigen. Positive colonies were selected and sub-cloned to yield one hybrid cell per well. Five positive clones were eventually generated and classified as 1C3, 1F6, 2D2, 3D11, and 4G1. Since all the five mAbs have similar characteristics (belonged to isotype IgG2a with kappa light chain), we randomly selected and evaluated 4G1. The specificity of 4G1 was then evaluated by IFA and Western blot assays. IFA results showed that green fluorescence was observed in PSV-infected PK15 cells and there was no fluorescence in mock infected-PK15 cells (Figure 4A). Western blot results revealed that mAb 4G1 was able to recognize a 77 kDa of MBP-tagged VP1 protein (Figure 4B) and a ~35 kDa VP1 protein in PK15 cells infected with PSV (Figure 4D). The cell lysate transformed with empty plasmid was set up as a positive control for MBP expression (~42 KDa) (Figure 4C).
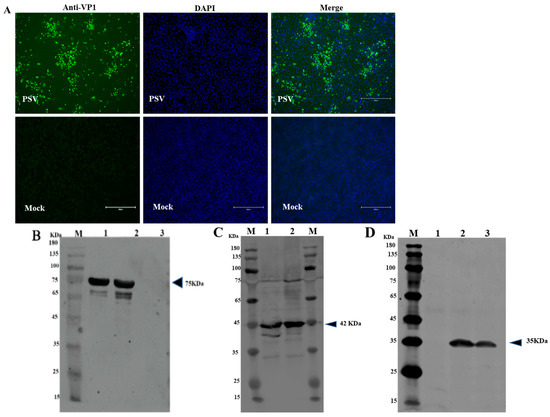
Figure 4. Identification of mAb against VP1 of PSV. (A) Identification of mAb against PSV-VP1 by IFA. PK15 cells infected with PSV at MOI of 0.01. IFA was performed using generated mAb against VP1. (B) Western blot analysis of the purified recombinant protein from E. Coli. Lane M, protein marker; Lane 1, 0.1 mg/mL purified MBP-tagged VP1-recombinant protein; Lane 2, 0.2 mg/mL MBP-tagged VP1-recombinant protein; Lane 3, purified MBP. (C) Western blot analysis of the expressed MBP in empty vector. Lane M, protein marker; Lane 1, sediment of bacterial cell lysate, Line 2, supernatant of bacterial cell lysate. (D) Western blot analysis of PK15 cells infected with PSV. Lane M, protein marker; Lane 1, mock cells; Lane 2, PK15 cells infected with PSV; Lane 3, ST cells infected with PSV.
7. Cell Susceptibility Test to the PSV Isolate
To evaluate the infection and replication ability of the isolated PSV strain, cell line susceptibility test was performed using nineteen cell lines derived from various species. The results revealed that the isolated PSV strain could only replicate in cell lines from swine origin and showed CPEs characterized by cell rounding, shrinking, and detachment at 24 h after infection, while other cell lines did not show CPEs. The evidence of productive infection of PSV in the swine cell lines was confirmed by the detection of viral VP1 protein expression using IFA with mAb 4G1 (Figure 5), suggesting that additional porcine primary factors might be necessary for PSV infection and replication.
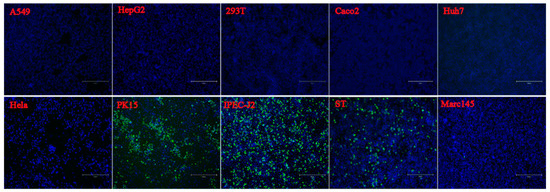
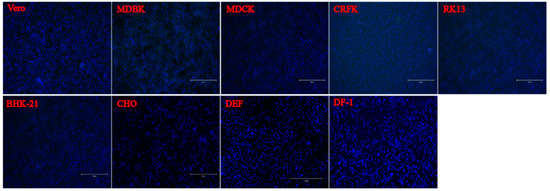
Figure 5. Susceptibility of PSV to cell lines derived from various species. Cell lines of Human (A549, HeLa, Huh-7, HepG2, Coca2 and 293T), Swine (PK15, IPEC-J2, and ST), Monkeys (Marc-145 and Vero), Bovine (MDBK), Canine (MDCK), Feline (CRFK), Rabbit (RK13), Hamster (BHK-21 and CHO), Duck (DEF) and Chicken (DF-1) were infected with PSV at an MOI of 0.01, and fixed at 24 hpi. Cell monolayers were determined by IFA using PSV mAb.
8. Establishment and Optimization of iELISA
IFA was used as a standard evaluating method for verifying seropositive and seronegative samples for iELISA optimization. In total 169 serum samples (72 positive and 97 negative samples), were collected from positive and negative swine by RT-PCR. The appropriate serum dilution for the IFA was determined by serial dilution (from 1:20 to 1:200) and found that the optimal dilution of serum was 1:100. The iELISA positive and negative sera were then confirmed by IFA, see the representative data in Figure 6A. To validate the serum samples, Western blot assay was further performed in PSV-infected PK15 cells by testing 48 samples (28 positive and 20 negative samples) randomly selected from the sera tested by IFA. It wasWe found that Western blot results were consistent with IFA, as shown in the representative data in Figure 6B,C. Interestingly, the VP1 protein with a molecular weight of 35 kDa was detected by the PSV-positive sera, but was not detected by the negative serum. Though PSV has four structural proteins, including VP1, VP2, VP3 and VP4, only VP1 was found to react with PSV-positive sera, implying that VP1 is the most abundant and strongest antigenic protein in the PSV particle.
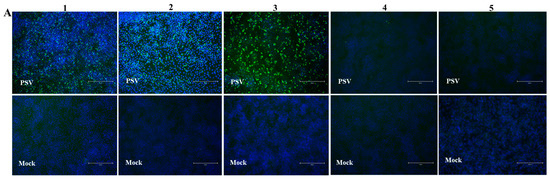
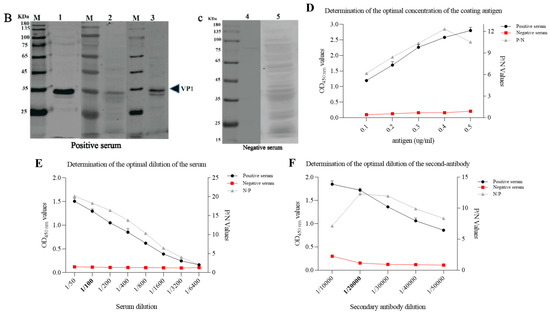
Figure 6. Optimization of reaction conditions of iELISA. (A) Verification of PSV-negative and positive swine sera by IFA. PK15 cells were inoculated with PSV at MOI of 0.01. IFA was performed using swine positive serum # 1, 2, and 3, and negative serum # 4 and 5. These are the representative sera of all the tested sera. (B,C) Confirmation of PSV positive and negative serum by Western blot. Protein lysates of PK15 cells infected with PSV were detected with PSV positive and negative swine serum respectively. (D–F) Optimization of the concentrations of coating antigen, serum dilutions and second antibody respectively.
To optimize the iELISA, the checkerboard titration was performed to determine the optimal concentration of the antigen and the serum. The results showed that the optimal antigen coating concentration was 0.2 µg/mL and the optimal dilution for serum was 1:100 (Figure 6D,E). Using this optimal dilution of coating protein and primary antibody, the optimal dilution of the HRP-conjugated goat anti-porcine IgG was determined to be 1:20,000, as shown in Figure 6F.
9. Determination of the Cut-Off Point and Evaluation of iELISA Specificity and Sensitivity
Seventy-five PSV-negative serum samples that were collected from swine negative to PSV by RT-PCR and confirmed by IFA and validated by Western blot assay, were used to determine the cut-off point. The cut-off point was specific to each assay and was based on the OD450nm of the 75 negative samples. Based on the mean OD450 nm plus 3 SD (0.161987 + 30.069591; n = 75; Max = 0.361; Min = 0.063), the cutoff value was determined to be 0.370 (Figure 7A). The negative-positive threshold was consequently set at 0.370, and serum samples with an absorbance of greater than 0.370 at OD450nm were considered PSV seropositive, and vice versa.
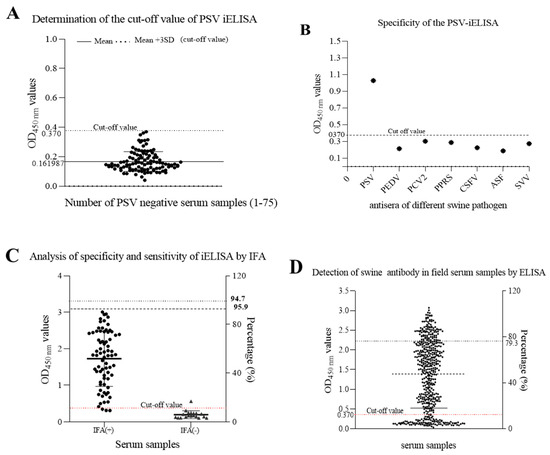
Figure 7. Detection of anti-PSV antibodies in swine serum by the developed iELISA. (A) seventy-five PSV-negative serum samples were tested using iELISA and the mean OD450 value of PSV-negative serum plus three standard deviations (SDs) were used to calculate the cutoff value. (B) Identification of iELISA specificity by cross-reaction test. Positive swine serum against PSV, PEDV, PCV2, PRRS, CSFV, ASFV and SVV were tested by iELISA. The average of OD450 values were calculated to determine the tested serum according to the cut-off value. (C) The sensitivity and specificity of the developed ELISA were assessed based on the IFA result. (D) Detection of anti-PSV antibodies from field swine serum samples by the developed iELISA.
To determine the specificity of this iELISA, porcine positive sera against other viruses, including PEDV, PCV2, PRRSV, CSFV, ASFV, and SVV, were examined. The results showed that there was no cross-reactivity of the newly developed iELISA with other porcine viruses, as shown in Figure 7B, and the average OD450 values of positive serum for PEDV, PCV2, PRRS, CSFV, ASFV, and SVV, were 0.213, 0.287, 0.302, 0.224, 0.187, and 0.273, respectively, indicating that the established iELISA was specific for the detection of PSV antibodies.
Furthermore, a total of 92 porcine sera collected from the field were tested in parallel using the iELISA and the IFA test. The results showed that positive and negative serum numbered 70 and 18 by IFA, but 73 and 19 by iELISA. In total, 70 samples were positive and 18 negatives in both cases, which represents a concurrence for 95.7%. Three positive samples in the iELISA test were negative in the IFA test, and one sample which was negative in the iELISA test have become positive in the IFA test. Hence, the sensitivity of iELISA was 95.9% among PSV-seropositive samples, and the specificity was 94.7% among PSV-seronegative samples using IFA as standard evaluation method.