Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 3 by Beatrix Zheng and Version 2 by Beatrix Zheng.
Both acute and chronic kidney diseases substantially contribute to the morbidities and mortality of patients worldwide. The existing therapeutics, which are mostly developed from synthetic sources, present some unexpected effects in patients, provoking researchers to explore potential novel alternatives. Natural products that have protective effects against various renal pathologies could be potential drug candidates for kidney diseases. Mangiferin is a natural polyphenol predominantly isolated from Mangifera indica and possesses multiple health benefits against various human ailments, including kidney disease.
- chronic kidney disease
- renoprotective
- kidney fibrosis
- oxidative stress
- inflammation
- mangiferin
1. Introduction
Kidney disease is a significant public health problem affecting more than 750 million people worldwide [1]. The burden of kidney disease varies substantially across the world, as does its detection and treatment. Kidney diseases are broadly classified into acute kidney injury (AKI) and chronic kidney disease (CKD). AKI denotes spontaneous kidney damage that generally lasts a few days to a week. The leading causes of AKI are damage to the kidney tissue by drugs or infections, leading to the blockage of urine (for example, a blockage can also be caused by kidney stones) [2]. If these conditions persist, kidney function decreases over time, leading to CKD. In the worst-case scenario, end-stage renal disease (ESRD), also known as kidney failure, can develop. There are many other factors or conditions involved in CKD, such as diabetes mellitus, glomerulonephritis, nephrosclerosis, and polycystic kidney disease [2]. There are many different causes of kidney disease, and sometimes the cause is unknown.
Renoprotective effects imply preserving the kidney structure or function in different pathological conditions. To date, there is no specific treatment option to completely inhibit the progression of AKI and CKD by preserving the integrity of architecture or function of kidneys. However, therapies in terms of kidney protection to confront the risk factors are worthy of consideration for maintaining the patient’s kidney function. A plethora of traditional treatments using natural products have shown a wide range of therapeutic windows for kidney diseases. Among these, one of the exemplary candidates is mangiferin (molecular formula: C19H18O11; systematic name: 1,3,6,7-tetrahydroxyxanthone C2-β-D-glucoside) (Figure 1), a naturally occurring glucoxilxanthone [3,4][3][4] derived from the various parts of Mangifera indica (Mango), including the leaves, fruits, flowers, seeds, roots, and stem bark [5]. In China, mangiferin is included in many traditional formulae that contain Iris domestica, Folium pyrrosiae, Gentiana scabra, and A nemarrhena asphodeloides. Moreover, this natural bioactive and polyhydroxy polyphenol element is enriched with several pharmacological beneficial effects without any known side effects [6].
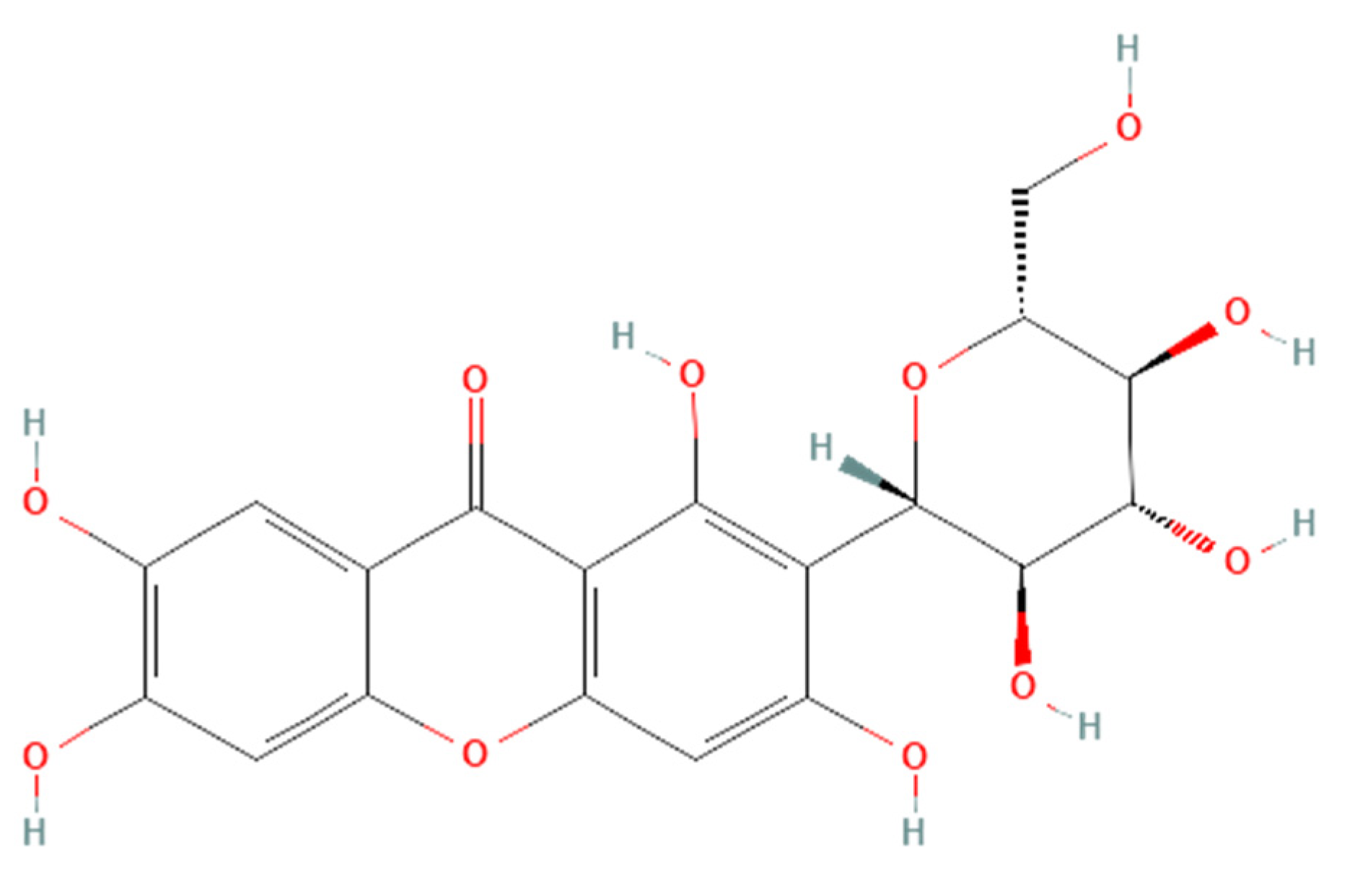 Figure 1. Structure of mangiferin (PubChem CID 5281647).
Figure 1. Structure of mangiferin (PubChem CID 5281647).Mangiferin is shown to have a renoprotective effect. Therefore, it has been extensively investigated regarding the beneficial effects of kidney diseases. Several reports support that mangiferin confers its renoprotective effects against AKI and CKD predominantly through protection against inflammation, scavenging of reactive oxygen species (ROS) in oxidative stress, anti-apoptotic and anti-fibrotic effects in renal intestinal fibrosis, preserving mitochondrial function, and reducing lipid peroxidation [7,8][7][8].
2. Pharmacological Effects of Mangiferin on Kidney Diseases
The pharmacological potential of mangiferin against several plausible factors such as oxidative stress, inflammation, fibrosis, and other pathologies associated with kidney diseases are summarized in this section (Figure 2, Figure 3 and Figure 4 and Table 1 and Table 2).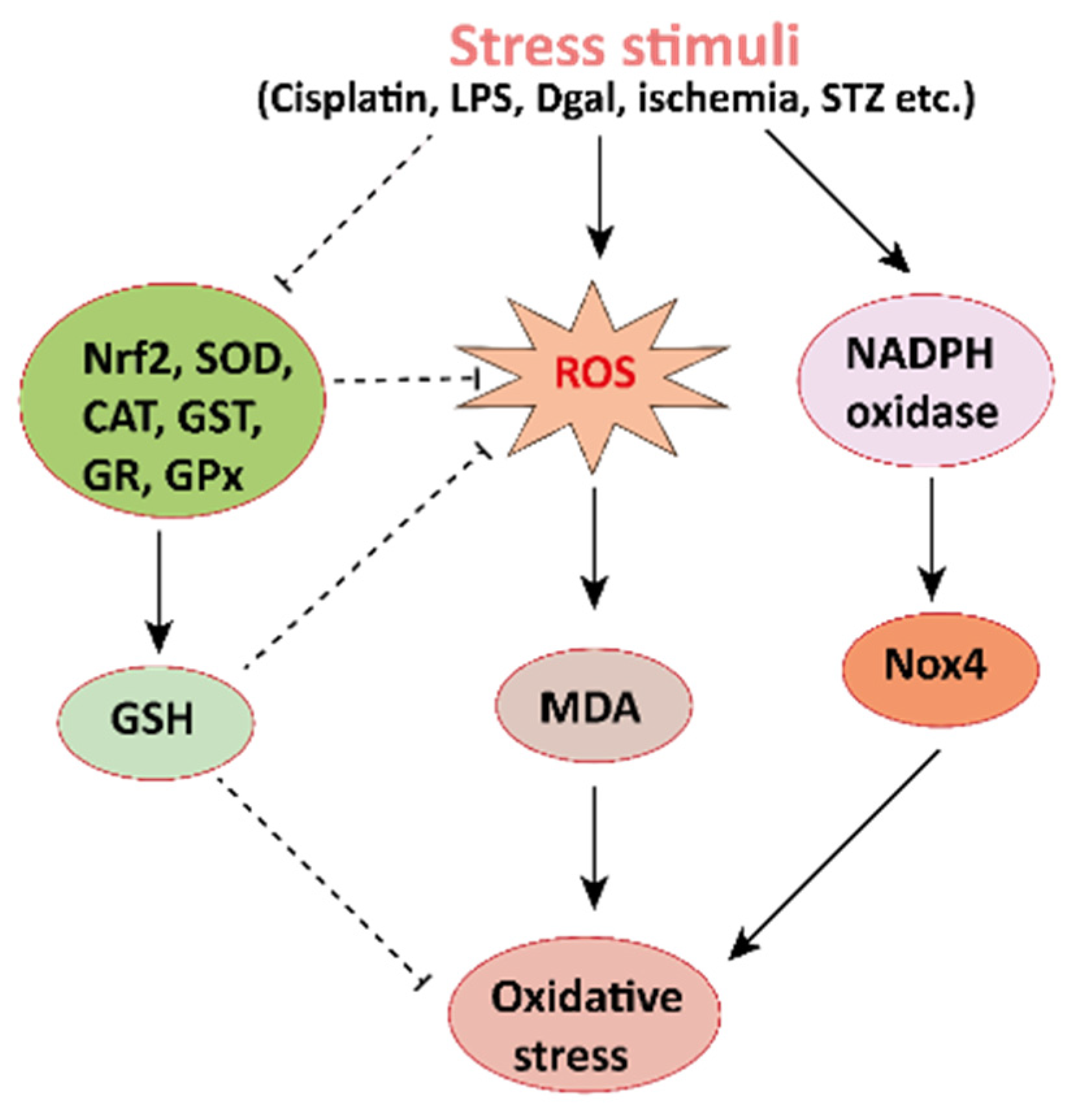 Figure 2. Mechanisms involved in the pathogenesis of oxidative stress in the kidney. NADPH oxidase is the main source of cellular ROS. Nox4 is an isoform of NADPH oxidase expressed in renal tubules that leads to oxidative stress (ROS and MDA) and damages the kidney. Stress stimuli, for instances cisplatin, STZ, ischemia result in decreased Nrf2 thus leading to oxidative stress. Reactive oxygen species, ROS; MDA, malondialdehyde; streptozotocin, STZ; NF-E2-related factor 2, Nrf2.
Figure 2. Mechanisms involved in the pathogenesis of oxidative stress in the kidney. NADPH oxidase is the main source of cellular ROS. Nox4 is an isoform of NADPH oxidase expressed in renal tubules that leads to oxidative stress (ROS and MDA) and damages the kidney. Stress stimuli, for instances cisplatin, STZ, ischemia result in decreased Nrf2 thus leading to oxidative stress. Reactive oxygen species, ROS; MDA, malondialdehyde; streptozotocin, STZ; NF-E2-related factor 2, Nrf2.
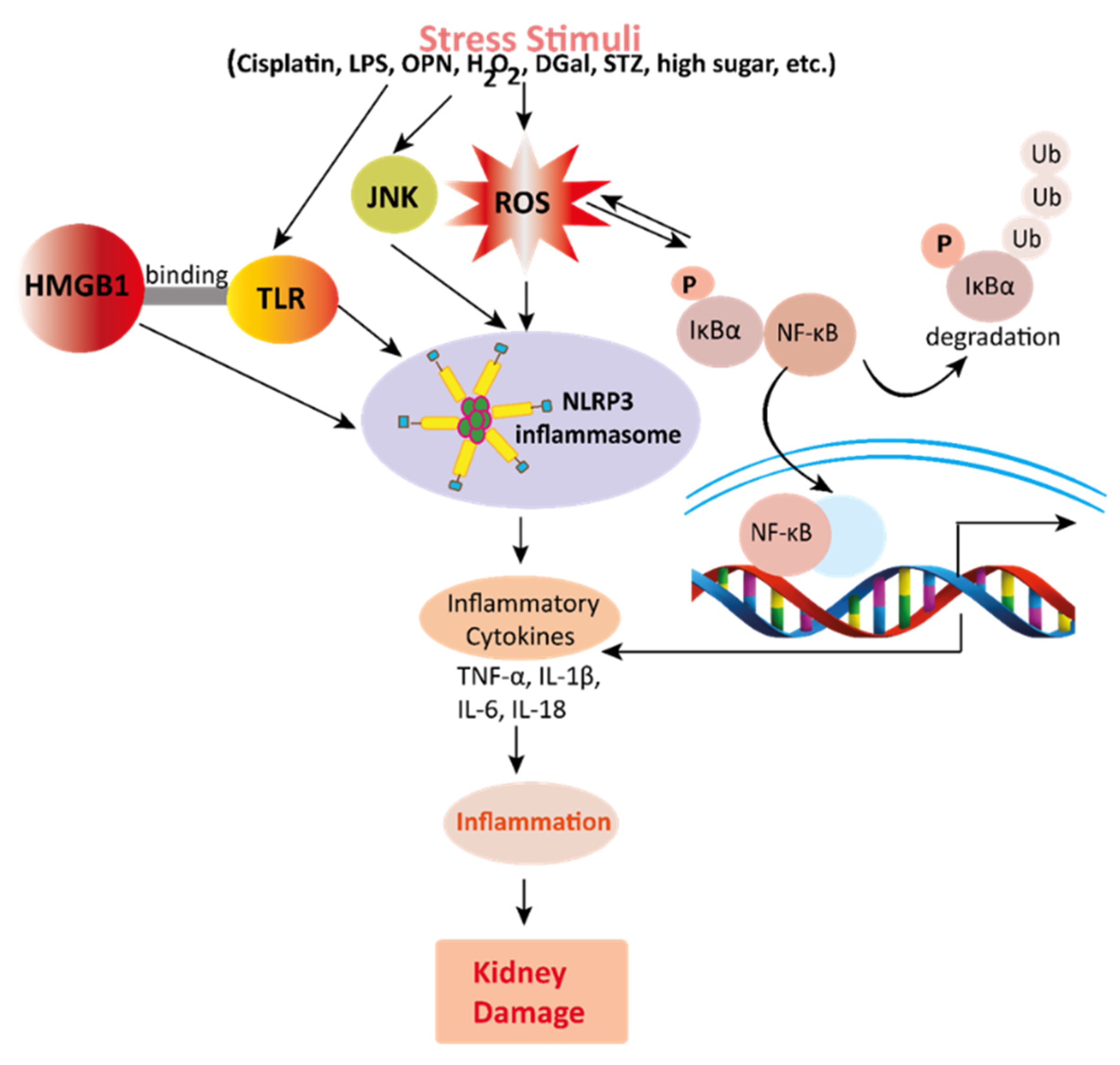 Figure 3. Mechanisms involved in the pathogenesis of inflammation in the kidney. Stress stimuli example for, cisplatin, DGal, STZ, LPS, OPN, high glucose, and H2O2 generate ROS in the kidney. Accumulation of ROS induces inflammation through the activation of the NLRP3 inflammasome. NLRP3 involves a multi-protein complex known as inflammasome, which triggers the NF-κB signaling pathway. HMGB1 protein is a late inflammation instigating compound which activates the NF-κB signaling pathway by binding with Toll-like receptors. The NF-κB pathway further promotes an inflammatory storm by releasing inflammatory cytokines (TNF-α, IL-1β, IL-6, IL-18), which ultimately leads to kidney damage. High-mobility group box 1; HMGB1, D (+) galactosamine; DGal, osteopontin; OPN, Streptozotocin; STZ, reactive oxygen species; ROS, IL-6; interleukin-6, IL-1β; Interleukin 1β, TNFα; Tumor Necrosis Factor-α.
Figure 3. Mechanisms involved in the pathogenesis of inflammation in the kidney. Stress stimuli example for, cisplatin, DGal, STZ, LPS, OPN, high glucose, and H2O2 generate ROS in the kidney. Accumulation of ROS induces inflammation through the activation of the NLRP3 inflammasome. NLRP3 involves a multi-protein complex known as inflammasome, which triggers the NF-κB signaling pathway. HMGB1 protein is a late inflammation instigating compound which activates the NF-κB signaling pathway by binding with Toll-like receptors. The NF-κB pathway further promotes an inflammatory storm by releasing inflammatory cytokines (TNF-α, IL-1β, IL-6, IL-18), which ultimately leads to kidney damage. High-mobility group box 1; HMGB1, D (+) galactosamine; DGal, osteopontin; OPN, Streptozotocin; STZ, reactive oxygen species; ROS, IL-6; interleukin-6, IL-1β; Interleukin 1β, TNFα; Tumor Necrosis Factor-α.
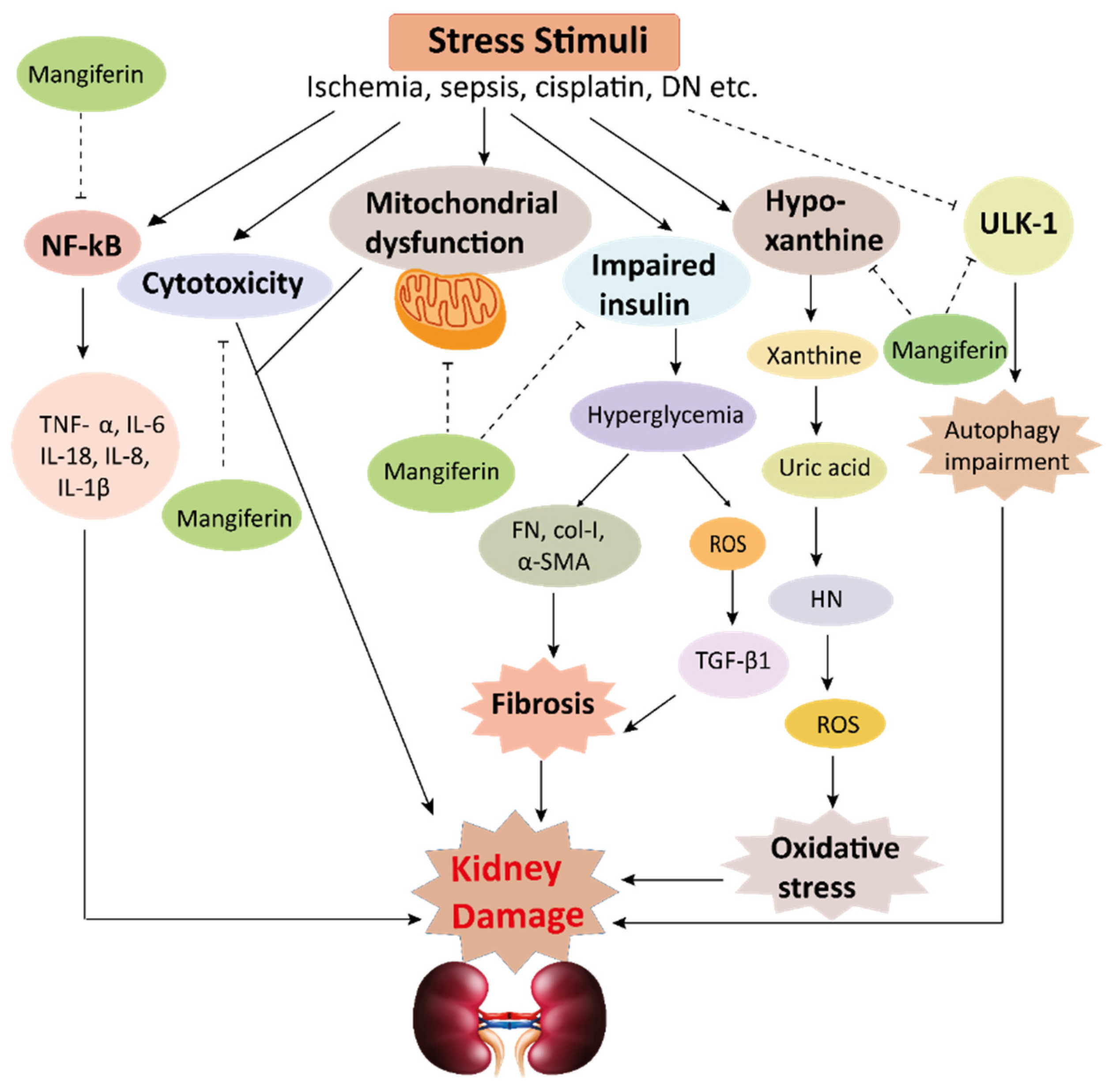 Figure 4. This schematic representation shows that stress stimuli example for, cisplatin, DN, Ischemia, and sepsis mediates various pathological conditions including cytotoxicity, oxidative stress, inflammation, fibrosis, autophagy dysfunction, and mitochondrial dysfunction. These ultimately lead to kidney damage. Stress stimuli activate the NF-kB signaling pathway which triggers the release of inflammatory cytokines (TNF-α, IL-6, IL-18, IL-8, and IL-1β) and decreases the action of ULK-1 thus autophagy impairment causes. Further col-1, FN, α-SMA causes accumulation of extracellular matrix (ECM) resulting in fibrosis. Mangiferin protects the kidney by suppressing the cascades of inflammatory pathways, oxidative stress, fibrosis, cytotoxicity, mitochondrial dysfunction, and autophagy impairment. ROS, reactive oxygen species; TNF-α, tumor necrosis factor-α; IL-1β, interleukin-1β; FN, fibronectin; α-SMA, α-smooth muscle actin; Col I, collagen I; TGF-β1, transforming growth factor, ULK-51, unc-51-like kinase; DN, diabetic nephropathy; HN, hyperuricemic nephropathy; NF-kB, nuclear factor-kappa B.
Figure 4. This schematic representation shows that stress stimuli example for, cisplatin, DN, Ischemia, and sepsis mediates various pathological conditions including cytotoxicity, oxidative stress, inflammation, fibrosis, autophagy dysfunction, and mitochondrial dysfunction. These ultimately lead to kidney damage. Stress stimuli activate the NF-kB signaling pathway which triggers the release of inflammatory cytokines (TNF-α, IL-6, IL-18, IL-8, and IL-1β) and decreases the action of ULK-1 thus autophagy impairment causes. Further col-1, FN, α-SMA causes accumulation of extracellular matrix (ECM) resulting in fibrosis. Mangiferin protects the kidney by suppressing the cascades of inflammatory pathways, oxidative stress, fibrosis, cytotoxicity, mitochondrial dysfunction, and autophagy impairment. ROS, reactive oxygen species; TNF-α, tumor necrosis factor-α; IL-1β, interleukin-1β; FN, fibronectin; α-SMA, α-smooth muscle actin; Col I, collagen I; TGF-β1, transforming growth factor, ULK-51, unc-51-like kinase; DN, diabetic nephropathy; HN, hyperuricemic nephropathy; NF-kB, nuclear factor-kappa B.
Table 1. In vivo renoprotective effects of mangiferin.
| Model Animals | Disease Inducing Agents | Mangiferin Dosages | ||
|---|---|---|---|---|
Table 2. In vitro renoprotective effects of mangiferin.
| Cell Lines | Model Drug | Effects of Mangiferin | Ref. | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Mangiferin | Effects of Mangiferin | Ref. | Dose | Route | Duration | Oxidative Stress | Inflammation | Fibrosis | Other Pathologies | ||||
| Dose | Duration | Oxidative Stress | Inflammation | Other Pathologies | |||||||||
| Mice | Cisplatin | 10, 20 and 40 mg/kg | orally | 21 days | ↓ ROS ↑ GSH, SOD, CAT, GST, GR, GPx |
↓ ROS↓ TNF-α, IL-1β, IL-6, IL-10, NF-κB |
− | ↓ Caspase-3 |
↑ GSH, SOD, CAT, GST, GR, GPx | [20 | − | ] | ↓ Caspase-3[9] |
| [ | Rats | Cisplatin | 20 and 40 mg/kg | i.p. | 10 days | ↓ ROS, MDA ↑ GSH, SOD, CAT |
↓ TNF-α, IL-6 | − | ↓Bax, caspase-3 ↑ Bcl-2↓ MAPK pathway |
[21] | [10] | ||
| Mice | STZ | 15, 30 and 60 mg/kg/day | orally | 4 Weeks | ↓ ROS, MDA | ||||||||
| Mice | |||||||||||||
| Ischemia | |||||||||||||
| 10, 30, and 100 mg/kg | |||||||||||||
| i.p. | |||||||||||||
| NKE cells | Cisplatin | 5–30 µM | 2 h | 20 | ] | [ | 9] | |||||||||||
| NKE Cells | NaF | 100–1000 μg/mL | ↓ ROS ↑ CAT, peroxidase and GHS |
− | − | [37] | [26] | |||||||||||
| ↑ SOD, CAT, andGSH-Px | ↓TNF-α, IL-6, IL-1β | ↓TGF-β1, FN, Col I, | ||||||||||||||||
| HRGEC | Cadmium | 75 μM | 24 h | ↓ ROS, MDA ↑ SOD, GSH, GR, GSH-Px |
↓ NF-kB, IL-6, IL-8 | ↓ Bax, Cytochrome C, Caspase ↑ Bcl-2 | and α-SMA |
↓Phosphorylation of PI3K and Akt | [38] | [[7] | ||||||||
| 27 | ] | Rats | STZ | 10, 20, 40, 60 and 80 mg/kg; | orally | |||||||||||||
| Mesangial Cells (SV40 MES 13) | 30 days | High glucose (25 mM) | 50 mg/kg | 48 hr | ↓ ROS | ↓ ROS, MDA ↑ SOD, CAT, GPX, GR |
↓ NF-kB, TNF-α | ↓ NOX4 | − | − | ↓ Caspase-3 ↑ Mitochondrial membranepotential | ↓ Cytochrome C |
[39] | [28 | ↓ Bax, Caspase-9, Caspase-3↑ Bcl-2↓ MAPK pathway |
] | [22] | [11] |
| Rats | STZ | 40 mg/kg | orally | 28 days | ↓ ROS ↑ CAT, GST, GS, GSH, GPx, SOD |
↓ NF-κB, TNF-α, VEGF, PKC |
↓ TGFβ1 | − | [23] | [12] | ||||||||
| Rats | STZ | 12.5, 25, or 50 mg/kg | orally | 12 weeks | − | − | − | ↑ AMPK ↓ mTOR ↑ pULK1 |
[24] | [13] | ||||||||
| Rats | STZ | 40 mg/kg/day | orally | 30 days | ↑ SOD, CAT, GPx and GSH ↓ ROS, MDA |
− | − | − | [25] | [14] | ||||||||
| Rats | STZ | 10 and 20 mg/kg | i.p | 28 days | ↓ ROS, MDA ↑ SOD, CAT |
− | − | − | [26] | [15] | ||||||||
| Rats | STZ | 15, 30, and 60 mg/kg | orally | 9 weeks | ↓ ROS, MDA, AGEs ↑ GSH |
− | − | − | [27] | [16] | ||||||||
| Mice | LPS | 20, 50, and 100 mg/kg | i.p. | 7 days | ↓ ROS, MDA ↑ SOD |
↓ NF-κB, HMGB1 | − | − | [28] | [17] | ||||||||
| Rats | LPS | 10, 50, 100 μM | 1 h | ↓ ROS, MDA | ↓ IL-1β, IL-18 and NLRP3 |
− | ↓ caspase-9 and caspase-3 |
[29] | [18] | |||||||||
| Mice | Uric acid | 50 mg/kg/day | orally | 7 days | ↓ ROS, XO ↑ SOD |
↓ IL-1b, IL-18 | − | − | [30] | [19] | ||||||||
| Mice | Uric Acid | 10, and 30 mg/kg | 7 days | ↓ ROS, MDA | − | − | − | [31] | [20] | |||||||||
| Rats and mice | Uric acid | 1.5–6.0 mg/kg | i.p. | 5 days | − | − | − | ↓ URAT1, OAT10, and GLUT9 | [32] | [21] | ||||||||
| Rats | OPN | 15, 30, and 60 mg/kg/day | orally | 9 weeks | − | ↓ TNF-α, COX-2, IL-1β, NF-κB, p65 |
↓ Col IV, α-SMA |
− | [33] | [22] | ||||||||
| Mice | tBHP | 75 mg/kg | orally | 2 weeks | ↑ SOD, CAT, GST, GRGPX | ↓ TNF-α, IL-6 and IL-1β |
− | ↓ Bax, caspase-8 caspase-3 ↑ Bcl-2 |
[34] | [23] | ||||||||
| Rats | DGal | 25 mg/kg | i.p. | 14 days | ↓ ROS, MDA ↑ CAT, GST, GS, GPx, SOD |
↓ NF-κB a, NO, TNF-α |
− | ↓ Bax, cytochrome c, caspase-9 and caspase-3 ↑ Bcl-2, Bcl-xL |
[35] | [24] | 30 min | − | ↓ TNF-α and IL-1β | − | ↓ Caspase-3 | [36] | [25] |
2.1. Effects of Mangiferin on Renal Oxidative Stress
Oxidative stress arises in the cells owing to an imbalance between production and accumulation of ROS, and subsequent inability to detoxify these reactive products (Figure 2) [40,41,42][29][30][31]. ROS is generated in the cells during the normal process of metabolism. Although optimum ROS level helps cell metabolism by cell-to-cell interactions, elevated ROS can cause extreme damage to cells and tissues [43][32]. The antioxidative defense mechanism of cells combined with an exogenous source of antioxidants is the key to minimizing the damages occurred by oxidative stress. Mangiferin, the natural bioactive compound has already been proven to have antioxidant properties. It may maintain a proper balance between ROS and antioxidants by reducing the level of intracellular ROS. It increases antioxidant activities in the kidney tissues, and subsequently stabilizes superoxide dismutase (SOD) activities, decreases uric acid synthesis, and improves antioxidant effects in mice and rats (Table 1).Cisplatin (cis-diamminedichloroplatinum II), is an anticancer drug that acts against several types of cancers by increasing intracellular ROS levels leading to oxidative stress, and reducing the antioxidant enzyme activities in the kidney epithelial (NKE) cells [20][9]. Importantly, mangiferin treatment reduces the kidney ROS levels, oxidative stress marker specifically MDA, and enhanced the antioxidant enzyme activities namely SOD, catalase, glutathione s-transferase (GST), glutathione peroxidase (GPx), and glutathione reductase (GR) are responsible for the degradation of ROS in the renal tissues both in vitro (NKE cells) [20][9] and in vivo (mice and rats) [20,21][9][10].
Several other drugs/chemical compounds cause oxidative damage by increasing ROS levels. Streptozotocin (STZ), a well-known poisonous substance forms ROS and MDA, resulting in induction of oxidative stress and reducing the levels of antioxidants [44][33]. Interestingly, mangiferin increased antioxidant enzymes as well as reduced ROS and MDA in STZ-induced diabetic mice and rats [7,22,23,24,25,26,27][7][11][12][13][14][15][16]. In diabetes, advance glycation end product (AGE) and xanthin oxidase plays an important role in inducing ROS; however, mangiferin treatment suppressed AGE generation and inhibited the activities of xanthin oxidase [22][11], indicating that mangiferin has the potential to reduce the levels of ROS in diabetic patients.
Mangiferin reduces the damage of antioxidants in lipopolysaccharide (LPS)-induced sepsis, thus, recovering sepsis-associated organ impairment [28][17] and increases antioxidant biomarkers in mouse kidney cells [29][18]. Besides, it has also been reported that mangiferin reduces intracellular MDA levels by acting as an effective ROS scavenger. Consistently, mangiferin ameliorated the oxidative stress in animal models with different ROS inducers namely uric acid, osteopontin, tert-Butylhydroperoxide (tBHP) and D(+)galactosamine (DGal) [30,31,32,33,34,35][19][20][21][22][23][24] as well as in different kidney cells [37,38,39][26][27][28], as briefly described in Table 1 and Table 2. Collectively, all these data from in vivo and in vitro studies indicated that mangiferin could be a potent therapeutic agent to combat oxidative stress in kidney diseases.
2.2. Effects of Mangiferin on Renal Inflammation
Renal inflammation is initiated by several factors including immune-mediated inflammatory mediators and a subsequent renal dysfunction or nephrotoxicity (Figure 3) [45,46,47,48,49][34][35][36][37][38]. Moreover, increased uric acid triggers renal inflammation through the c-Jun N-terminal kinases (JNK) signaling pathway and nucleotide-binding oligomerization domain (NOD)-, leucine-rich repeat (LRR)- and pyrin domain-containing protein (NLRP) 3 inflammasome. NLRP3 inflammasome stimulates pro-inflammatory cytokines interleukin-1β (IL-1β) and IL-18 production and excretion, thus promoting renal injuries and disease in mice such as hyperuricemic nephropathy (HN) [30][19]. In connection with these, recent studies demonstrated the anti-inflammatory effects of mangiferin in kidney tissues and cells (Table 1 and Table 2). Mechanistically, mangiferin shows its anti-inflammatory effects by inhibiting the activation of both the JNK pathway and NLRP3 inflammasome and minimizing urate [29][18]. It has also demonstrated that mangiferin treatment preserved the glomerular and tubular structures of kidneys in mice [30][19]. Cisplatin manifests nephrotoxicity in the liver with deleterious effects in the renal tissues. Cisplatin exposure escalates proinflammatory cytokines (tumor necrosis factor-α; TNF-α, IL-1β, IL-6, IL-10) and nuclear factor-kappa β (NF-κB) in the nuclear fraction of the renal tissue in rats and mice; however, mangiferin treatment attenuated the level of all these cytokines [20,21][9][10].
Studies demonstrated that high mobility group box 1 (HMGB1), a protein that binds to toll-like receptors (TLR), activates NF-κB inflammatory signaling pathways inducing the release of inflammatory factors; hence, it contributes to an inflammatory storm in LPS-treated mice, and interestingly, mangiferin prevented the activation of this signaling pathway [28][17]. Moreover, simultaneous administration of mangiferin reduced cisplatin-instigated nephrotoxicity in vitro and in vivo by preventing the nuclear translocation of NF-κB and proinflammatory cytokines. Blocking NF-κB pathway by mangiferin defeats the NF-κB cascade pathway of inflammation in rats and mice [20,21][9][10]. In contrast, tBHP instigated inflammation [34][23], as well as STZ induced-diabetic nephropathy (DN) [7,22,23,24,25,26,27][7][11][12][13][14][15][16] were ameliorated by mangiferin. Moreover, DGal advances inflammation by prompting NF-κB and TNFα in rats in renal tissue, and osteopontin (OPN), a proinflammatory cytokine, assists in inflammatory gene expression. Mangiferin preserves renal function appositely against NLRP3 inflammasome [33][22] and lessens inflammation by obstructing DGal [35][24] and OPN [33][22]. In diabetic nephropathy, high glucose generates an inflammatory response characterized by activating the NF-κB pathway, TNF-α, and IL-1β in the renal tissue of diabetic rats, and mangiferin was able to reduce the burden of inflammation in DN [39][28]. Moreover, mangiferin inhibits renal ischemia-reperfusion damages by blocking inflammatory agents [36][25] and preserves renal function against cadmium-induced eukaryotic cell death via the NF-κB signaling pathways [38][27]. All data suggest that mangiferin has the ability to combat inflammation in the kidneys.
2.3. Effects of Mangiferin on Renal Fibrosis
Renal inflammation is initiated by several factors including immune-mediated inflammatory mediators and a subsequent renal dysfunction or nephrotoxicity (Figure 3) [45,46,47,48,49][34][35][36][37][38]. Moreover, increased uric acid triggers renal inflammation through the c-Jun N-terminal kinases (JNK) signaling pathway and nucleotide-binding oligomerization domain (NOD)-, leucine-rich repeat (LRR)- and pyrin domain-containing protein (NLRP) 3 inflammasome. NLRP3 inflammasome stimulates pro-inflammatory cytokines interleukin-1β (IL-1β) and IL-18 production and excretion, thus promoting renal injuries and disease in mice such as hyperuricemic nephropathy (HN) [30][19]. In connection with these, recent studies demonstrated the anti-inflammatory effects of mangiferin in kidney tissues and cells (Table 1 and Table 2). Mechanistically, mangiferin shows its anti-inflammatory effects by inhibiting the activation of both the JNK pathway and NLRP3 inflammasome and minimizing urate [29][18]. It has also demonstrated that mangiferin treatment preserved the glomerular and tubular structures of kidneys in mice [30][19]. Cisplatin manifests nephrotoxicity in the liver with deleterious effects in the renal tissues. Cisplatin exposure escalates proinflammatory cytokines (tumor necrosis factor-α; TNF-α, IL-1β, IL-6, IL-10) and nuclear factor-kappa β (NF-κB) in the nuclear fraction of the renal tissue in rats and mice; however, mangiferin treatment attenuated the level of all these cytokines [20,21][9][10].
Studies demonstrated that high mobility group box 1 (HMGB1), a protein that binds to toll-like receptors (TLR), activates NF-κB inflammatory signaling pathways inducing the release of inflammatory factors; hence, it contributes to an inflammatory storm in LPS-treated mice, and interestingly, mangiferin prevented the activation of this signaling pathway [28][17]. Moreover, simultaneous administration of mangiferin reduced cisplatin-instigated nephrotoxicity in vitro and in vivo by preventing the nuclear translocation of NF-κB and proinflammatory cytokines. Blocking NF-κB pathway by mangiferin defeats the NF-κB cascade pathway of inflammation in rats and mice [20,21][9][10]. In contrast, tBHP instigated inflammation [34][23], as well as STZ induced-diabetic nephropathy (DN) [7,22,23,24,25,26,27][7][11][12][13][14][15][16] were ameliorated by mangiferin. Moreover, DGal advances inflammation by prompting NF-κB and TNFα in rats in renal tissue, and osteopontin (OPN), a proinflammatory cytokine, assists in inflammatory gene expression. Mangiferin preserves renal function appositely against NLRP3 inflammasome [33][22] and lessens inflammation by obstructing DGal [35][24] and OPN [33][22]. In diabetic nephropathy, high glucose generates an inflammatory response characterized by activating the NF-κB pathway, TNF-α, and IL-1β in the renal tissue of diabetic rats, and mangiferin was able to reduce the burden of inflammation in DN [39][28]. Moreover, mangiferin inhibits renal ischemia-reperfusion damages by blocking inflammatory agents [36][25] and preserves renal function against cadmium-induced eukaryotic cell death via the NF-κB signaling pathways [38][27]. All data suggest that mangiferin has the ability to combat inflammation in the kidneys.
2.4. Effects of Mangiferin against Other Kidney Pathologies
ROS activates mitogen-activated protein kinases (MAPK) pathways along with JNK, p38 kinases, and extracellular signal-regulated kinases (ERK) by inducing cellular stress in rats [7]. The downstream pathway of MAPK involves three essential serine kinase proteins, including JNK, ERK1/2, and p38. These proteins assist in apoptosis and cell death. Importantly, mangiferin treatment reduced ROS generation and subsequently alter the MAPK pathway [21][10]. In contrast, proapoptotic (Bax, Bad, etc.) and anti-apoptotic (Bcl-2, Bcl-xl) proteins are entangled in apoptosis in STZ persuaded diabetic rats, which is attenuated by mangiferin treatment [22][11]. TNF-α activates caspase 8, while caspase 8 and 3 play a significant role to ensure programmed cell death that is apoptosis. Also, phosphorylation of JNK protein executes apoptosis and mangiferin treatment successfully prevents apoptosis in kidney tissues [22][11]. DGal, a toxic component, upregulates caspase 3/9 and changes the reciprocal regulation of Bcl-2 family proteins and these proteins govern permeabilization of outer membrane mitochondria. Bax and Bad proteins initiate apoptosis, release cytochrome c into the cytosol, and gradually activate the pro-apoptotic caspase cascade. Anti-apoptotic proteins Bcl-2 and Bcl-xL suppress Bax and Bak oligomer [35][24]. AMP-activated protein kinase (AMPK), a serine protein kinase that responds to cellular stress is activated when cellular energy is insufficient, thus inhibits mammalian target of rapamycin complex (mTOR) activity and increases autophagy. Autophagy refers to the degradation associated with the clearance of injured proteins and organelles initiated by activating the unc-51-like kinase 1 (ULK1) complex. ULK1 complex is prevented by mTOR activities AMPK phosphorylation decreases mTOR phosphorylation and manifests a nephroprotective effect by regulating MAPK. Mangiferin treatment was proven to have beneficial effects on these molecular machineries [24][13]. Moreover, pre-treatment with mangiferin exerts uricosuric action in hyperuricemic rats associated with obstruction of urate reabsorption through down-regulation of the mRNA and protein expressions of urate transporters in renal cells [32][21]. It also damages apoptotic proteins such as p53 and Nrf-2 related signaling cascades, reduces caspase and mitochondrial dysfunction [20,22][9][11]. Furthermore, it rehabilitates the altered Bax/Bcl-2 ratio by reducing mitochondrial dysfunction, releasing cytochrome C in the cytoplasm from the mitochondria [30][19], and diminishing Bax proteins in DGal revealed rats [35][24]. Mangiferin exerts beneficial effects on these different molecular cascades.
References
- Kassebaum, N.J.; Arora, M.; Barber, R.M.; Brown, J.; Carter, A.; Casey, D.C.; Charlson, F.J.; Coates, M.M.; Coggeshall, M.; Cornaby, L.; et al. Global, regional, and national disability-adjusted life-years (DALYs) for 315 diseases and injuries and healthy life expectancy (HALE), 1990–2015: A systematic analysis for the Global Burden of Disease Study 2015. Lancet 2016, 388, 1603–1658.
- Chawla, L.S.; Eggers, P.W.; Star, R.A.; Kimmel, P.L. Acute Kidney Injury and Chronic Kidney Disease as Interconnected Syndromes. N. Engl. J. Med. 2014, 371, 58–66.
- Matkowski, A.; Kus, P.; Goralska, E.; Wozniak, D. Mangiferin—A bioactive xanthonoid, not only from mango and not just antioxidant. Mini-Rev. Med. Chem. 2013, 13, 439–455.
- Sekar, M. Molecules of interest-mangiferin—A review. Annu. Res. Rev. Biol. 2015, 5, 307–320.
- Jyotshna; Khare, P.; Shanker, K. Mangiferin: A review of sources and interventions for biological activities. BioFactors 2016, 42, 504–514.
- Benard, O.; Chi, Y. Medicinal properties of mangiferin, structural features, derivative synthesis, pharmacokinetics and biological activities. Mini-Rev. Med. Chem. 2015, 15, 582–594.
- Song, Y.; Liu, W.; Tang, K.; Zang, J.; Li, D.; Gao, H. Mangiferin alleviates renal interstitial fibrosis in streptozotocin-induced diabetic mice through regulating the PTEN/PI3K/Akt signaling pathway. J. Diabetes Res. 2020, 2020, 9481720.
- Saha, S.; Sadhukhan, P.; Sil, P.C. Mangiferin: A xanthonoid with multipotent anti-inflammatory potential. BioFactors 2016, 42, 459–474.
- Sadhukhan, P.; Saha, S.; Dutta, S.; Sil, P.C. Mangiferin ameliorates cisplatin induced acute kidney injury by upregulating Nrf-2 via the activation of PI3K and exhibits synergistic anticancer activity with cisplatin. Front. Pharmacol. 2018, 9, 638.
- Sahu, A.K.; Verma, V.K.; Mutneja, E.; Malik, S.; Nag, T.C.; Dinda, A.K.; Arya, D.S.; Bhatia, J. Mangiferin attenuates cisplatin-induced acute kidney injury in rats mediating modulation of MAPK pathway. Mol. Cell. Biochem. 2019, 452, 141–152.
- Pal, P.B.; Sinha, K.; Sil, P.C. Mangiferin attenuates diabetic nephropathy by inhibiting oxidative stress mediated signaling cascade, TNFα related and mitochondrial dependent apoptotic pathways in streptozotocin-induced diabetic rats. PLoS ONE 2014, 9, e115364.
- Sekar, V.; Mani, S.; Malarvizhi, R.; Barathidasan, R.; Vasanthi, H.R. Positive interaction of mangiferin with selected oral hypoglycemic drugs: A therapeutic strategy to alleviate diabetic nephropathy in experimental rats. Mol. Biol. Rep. 2020, 47, 4465–4475.
- Wang, X.; Gao, L.; Lin, H.; Song, J.; Wang, J.; Yin, Y.; Zhao, J.; Xu, X.; Li, Z.; Li, L. Mangiferin prevents diabetic nephropathy progression and protects podocyte function via autophagy in diabetic rat glomeruli. Eur. J. Pharmacol. 2018, 824, 170–178.
- Sellamuthu, P.S.; Arulselvan, P.; Kamalraj, S.; Fakurazi, S.; Kandasamy, M. Protective nature of mangiferin on oxidative stress and antioxidant status in tissues of streptozotocin-induced diabetic rats. ISRN Pharmacol. 2013, 2013, 750109.
- Muruganandan, S.; Gupta, S.; Kataria, M.; Lal, J.; Gupta, P. Mangiferin protects the streptozotocin-induced oxidative damage to cardiac and renal tissues in rats. Toxicology 2002, 176, 165–173.
- Liu, Y.-W.; Zhu, X.; Zhang, L.; Lu, Q.; Wang, J.-Y.; Zhang, F.; Guo, H.; Yin, J.-L.; Yin, X.-X. Up-regulation of glyoxalase 1 by mangiferin prevents diabetic nephropathy progression in streptozotocin-induced diabetic rats. Eur. J. Pharmacol. 2013, 721, 355–364.
- Zhang, D.; Han, S.; Zhou, Y.; Qi, B.; Wang, X. Therapeutic effects of mangiferin on sepsis-associated acute lung and kidney injuries via the downregulation of vascular permeability and protection of inflammatory and oxidative damages. Eur. J. Pharm. Sci. 2020, 152, 105400.
- He, L.; Peng, X.; Zhu, J.; Chen, X.; Liu, H.; Tang, C.; Dong, Z.; Liu, F.; Peng, Y. Mangiferin attenuate sepsis-induced acute kidney injury via antioxidant and anti-inflammatory effects. Am. J. Nephrol. 2014, 40, 441–450.
- Li, X.; Yan, Z.; Carlström, M.; Tian, J.; Zhang, X.; Zhang, W.; Wu, S.; Ye, F. Mangiferin ameliorates hyperuricemic nephropathy which is associated with downregulation of AQP2 and increased urinary uric acid excretion. Front. Pharmacol. 2020, 11, 49.
- Qin, Z.; Wang, S.; Lin, Y.; Zhao, Y.; Yang, S.; Song, J.; Xie, T.; Tian, J.; Wu, S.; Du, G. Antihyperuricemic effect of mangiferin aglycon derivative J99745 by inhibiting xanthine oxidase activity and urate transporter 1 expression in mice. Acta Pharm. Sin. B 2018, 8, 306–315.
- Yang, H.; Gao, L.; Niu, Y.; Zhou, Y.; Lin, H.; Jiang, J.; Kong, X.; Liu, X.; Li, L. Mangiferin inhibits renal urate reabsorption by modulating urate transporters in experimental hyperuricemia. Biol. Pharm. Bull. 2015, 38, 1591–1598.
- Zhu, X.; Cheng, Y.-Q.; Du, L.; Li, Y.; Zhang, F.; Guo, H.; Liu, Y.-W.; Yin, X.-X. Mangiferin attenuates renal fibrosis through down-regulation of osteopontin in diabetic rats. Phyther. Res. 2015, 29, 295–302.
- Saha, S.; Mahalanobish, S.; Dutta, S.; Sil, P.C. Mangiferin ameliorates collateral neuropathy in t BHP induced apoptotic nephropathy by inflammation mediated kidney to brain crosstalk. Food Funct. 2019, 10, 5981–5999.
- Ghosh, M.; Das, J.; Sil, P.C. D(+) galactosamine induced oxidative and nitrosative stress-mediated renal damage in rats via NF-κB and inducible nitric oxide synthase (iNOS) pathways is ameliorated by a polyphenol xanthone, mangiferin. Free Radic. Res. 2012, 46, 116–132.
- Wang, B.; Wan, J.; Gong, X.; Kuang, G.; Cheng, X.; Min, S. Mangiferin attenuates renal ischemia-reperfusion injury by inhibiting inflammation and inducing adenosine production. Int. Immunopharmacol. 2015, 25, 148–154.
- Samadarsi, R.; Dutta, D. Anti-oxidative effect of mangiferin-chitosan nanoparticles on oxidative stress-induced renal cells. Int. J. Biol. Macromol. 2020, 151, 36–46.
- Rajendran, P.; Rengarajan, T.; Nishigaki, Y.; Palaniswami, R.; Nishigaki, I. In vitro studies on mangiferin protection against cadmium-induced human renal endothelial damage and cell death via the MAP kinase and NF-κB pathways. J. Recept. Signal Transduct. 2016, 36, 57–66.
- Xu, G.-K.; Sun, C.-Y.; Qin, X.-Y.; Han, Y.; Li, Y.; Xie, G.-Y.; Min-Jian, Q. Effects of ethanol extract of Bombax ceiba leaves and its main constituent mangiferin on diabetic nephropathy in mice. Chin. J. Nat. Med. 2017, 15, 597–605.
- Kao, M.P.; Ang, D.S.; Pall, A.; Struthers, A.D. Oxidative stress in renal dysfunction: Mechanisms, clinical sequelae and therapeutic options. J. Hum. Hypertens. 2010, 24, 1–8.
- Gyurászová, M.; Gurecká, R.; Bábíčková, J.; Tóthová, Ľ. Oxidative stress in the pathophysiology of kidney disease: Implications for noninvasive monitoring and identification of biomarkers. Oxid. Med. Cell. Longev. 2020, 2020, 5478708.
- Uddin, M.J.; Kim, E.H.; Hannan, M.A.; Ha, H. Pharmacotherapy against oxidative stress in chronic kidney disease: Promising small molecule natural products targeting nrf2-ho-1 signaling. Antioxidants 2021, 10, 258.
- Schieber, M.; Chandel, N.S. ROS function in redox signaling and oxidative stress. Curr. Biol. 2014, 24, R453.
- Kwon, G.; Uddin, M.J.; Lee, G.; Jiang, S.; Cho, A.; Lee, J.H.; Lee, S.R.; Bae, Y.S.; Moon, S.H.; Lee, S.J.; et al. A novel pan-Nox inhibitor, APX-115, protects kidney injury in streptozotocin-induced diabetic mice: Possible role of peroxisomal and mitochondrial biogenesis. Oncotarget 2017, 8, 74217–74232.
- Wang, T.; Fu, X.; Chen, Q.; Patra, J.K.; Wang, D.; Wang, Z.; Gai, Z. Arachidonic acid metabolism and kidney inflammation. Int. J. Mol. Sci. 2019, 20, 3683.
- Uddin, M.J.; Jeong, J.; Pak, E.S.; Ha, H. CO-releasing molecule-2 prevents acute kidney injury through suppression of ROS-Fyn-ER stress signaling in mouse model. Oxid. Med. Cell. Longev. 2021, 2021, 9947772.
- Uddin, M.J.; Dorotea, D.; Pak, E.S.; Ha, H. Fyn kinase: A potential therapeutic target in acute kidney injury. Biomol. Ther. 2020, 28, 213–221.
- Uddin, M.J.; Pak, E.S.; Ha, H. Carbon monoxide releasing molecule-2 protects mice against acute kidney injury through inhibition of ER stress. Korean J. Physiol. Pharmacol. 2018, 22, 567–575.
- Moni, A.; Iqbal, A.; Uddin, M. Resveratrol attenuates inflammation through tristetraprolin expression in human hepatocytes. J. Adv. Biotechnol. Exp. Ther. 2018, 1, 78–82.
More