Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Catherine Yang and Version 1 by Valeria Gasperi.
Niacin (also known as “vitamin B3” or “vitamin PP”) includes two vitamers (nicotinic acid and nicotinamide) giving rise to the coenzymatic forms nicotinamide adenine dinucleotide (NAD) and nicotinamide adenine dinucleotide phosphate (NADP).
- central nervous system
- niacin
1. Introduction
Niacin (also known as “vitamin B3” or “vitamin PP”) is the generic descriptor for two vitamers, nicotinic acid (pyridine-3-carboxylic acid) and nicotinamide (nicotinic acid amide), that give rise to the biologically active coenzymes, nicotinamide adenine dinucleotide (NAD) and its phosphate analog, the nicotinamide adenine dinucleotide phosphate (NADP) [1] (Figure 1). The two coenzymes take part in redox reactions crucial for energy production: in particular, the pyridinic ring can accept and donate a hydride ion (:H−, the equivalent of a proton and two electrons), thus acting as an electron carrier. Nonetheless, NAD and NADP play different metabolic roles in the cytosol: the NADH/NAD+ ratio is small (about 8 × 10−4), thus favoring oxidative catabolism, whereas the NADPH/NADP+ ratio is higher (about 75), thus providing a strongly reducing environment for biosynthetic reactions [2,3][2][3].
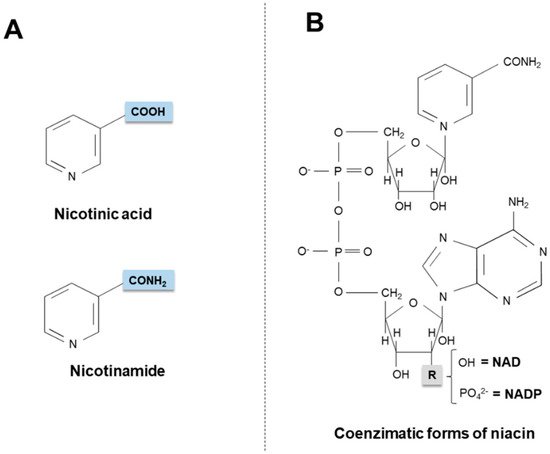
Figure 1. Chemical structures of niacin vitamers (A) and active coenzymatic forms (B). NAD: nicotinamide adenin dinucleotide. NADP: nicotinamide adenin dinucleotide phosphate.
Maintenance of the intracellular NAD pool is not only important to fuel redox metabolism, but also to support NAD-dependent, non-redox signaling pathways. NAD is indeed a substrate of ADP-ribosyltransferases that catalyze ADP-ribose transfer reactions, thus breaking down NAD to nicotinamide and ADP-ribosyl products, which play a key role in cellular signaling cascades regulating gene expression, cell cycle progression, insulin secretion, DNA repair, apoptosis and aging [4,5,6][4][5][6]. Finally, NAD has also been recognized as an endogenous agonist of purinergic P2Y1 and P2Y11 membrane subtype receptors, through which it inhibits neurotransmission in visceral smooth muscles [7] and activates immune cells [8[8][9],9], respectively.
2. Niacin Sources
Humans obtain niacin from both endogenous and exogenous sources. Only 2% of dietary tryptophan (Trp) is converted into niacin via a multistep pathway (see in next sections), occurring mainly in the liver [10]. Diet provides the vitamin as nicotinic acid, nicotinamide and Trp, as well as the active coenzymatic forms of niacin.
2.1. Exogenous Sources
Niacin is found in animal and vegetable foods. In meat and fish, the vitamin is present as NAD(P), whose amounts are higher in unprepared foods compared to processed foods (enzymatic hydrolysis of the coenzymes can occur during food preparation).
In mature cereal grains (particularly in corn), niacin is largely present as niacin-glycoside and, in a minor proportion, peptide-bound niacin, compounds collectively termed “niacinogens” [11]. When complexed in niacinogens, niacin is poorly available (only ~ 30%), as intestinal enzymes are not able to free niacin; nonetheless, alkali treatment of the grain increases niacin bioavailability [11].
Once ingested, free niacin can be adsorbed in the stomach, although the small intestine absorbs it faster. The mechanism of transport across the enterocyte brush border membrane is not fully clarified yet. Several transporters, indeed, appear to be involved in intestinal niacin uptake; among them, the most common are the human organic anion transporter-10 (hOAT-10, a proton-driven carrier that also mediates the transport of urate and p-aminohippurate) [12], responsible for niacin uptake at physiological concentrations [13], and the sodium-coupled monocarboxylate transporter (SMCT1 or SLC5A8, a transporter for lactate, pyruvate and short-chain fatty acids), specifically active at high pharmacological doses of nicotinic acid [14,15][14][15].
NAD and NADP are quickly hydrolyzed, by intestinal mucosa and liver glycohydrolases, to nicotinamide that is subsequently transported to tissues, where it is converted into coenzymatic forms as necessary. It seems noteworthy that nicotinamide moves freely into or out of the brain [16] and, as discussed in the next sections, such a property has important neurobiological implications.
2.2. Endogenous Synthesis
Starting from dietary Trp, niacin is synthesized via the kynurenine pathway (KP) (Figure 2), occurring mainly in the liver and, to a lesser extent, in extrahepatic tissues (especially upon immune cell activation) [17,18,19][17][18][19].
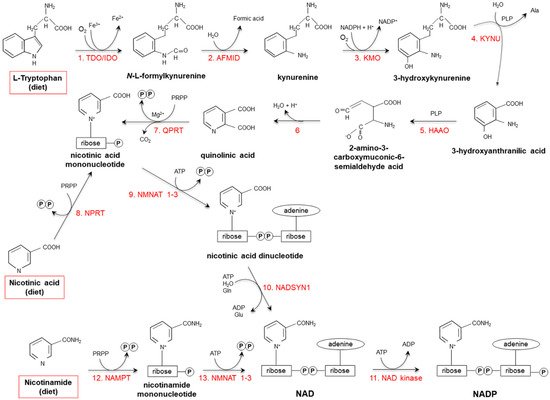
Figure 2. De novo synthesis of NAD(P) from tryptophan, nicotinamide and nicotinic acid. (1) Two iron porphyrin metalloproteins, tryptophan 2,3 dioxygenase (TDO, in the liver) and indolamine-pyrrole 2-3 dioxygenase (IDO, in extrahepatic tissues), oxidize the pyrrole moiety of Tryptophan (Trp), thus forming N-L-formylkynurenine. (2) Arylformamidase (AFMID) hydrolytically removes the formyl group producing kynurenine and is then (3) hydroxylated to 3-hydroxykynurenine by kynurenine-3 monooxygenase (KMO), a mitochondrial flavo-enzyme that uses O2 as a substrate and NADPH as a cofactor. The action of (4) kynureninase B (KYNU, a vitamin B6-dependent enzyme) and (5) 3-hydroxyanthranilic dioxygenase (HAAO, a nonheme iron-dependent dioxygenase) leads to production of 2-amino-3-carboxymuconic-6-semialdehyde acid, an unstable product that (6) spontaneously condensates and rearranges to form quinolinic acid; then, (7) quinolinic acid is decarboxylated and converted to nicotinic acid mononucleotide by quinolinic acid phosphoribosyltransferase (QPRT). Nicotinic acid mononucleotide is also produced through the “salvage pathway”, via the action of (8) nicotinic acid phosphoribosyltransferase (NPRT). The subsequent action of (9) nicotinamide/nicotinic acid-mononucleotide-adenylyltransferases (NMNAT1-3) and (10) NAD synthetase (NADSYN1) leads to the generation of NAD, which is then (11) phosphorylated to produce NADP. NAD can also derive directly from nicotinamide through the action of (12) nicotinamide phosphoribosyltransferase (NAMPT) and (13) nicotinamide/nicotinic acid-mononucleotide-adenylyltransferase (NMNAT1-3). Red frames: dietary precursors of NAD(P). Ala: alanine; Gln: glutamine; Glu: glutamate; PLP pyridoxal phosphate; PRPP: 5-phosphoribosyl-1- pyrophosphate.
Tryptophan 2,3 dioxygenase (TDO), catalyzing the first reaction, is the rate-limiting enzyme. Several nutritional, hormonal and physio-pathological factors affect the efficiency of this anabolic pathway. Deficiencies of vitamin B6, riboflavin, iron and heme (all essential cofactors for specific enzymes), as well as of vitamin B1 and Trp itself, slow the reaction rate [18,20][18][20]. Overall: (i) a protein-enriched diet (particularly, consumption of foods with high concentrations of leucine, such as maize or sorghum) decreases niacin biosynthesis; (ii) unsaturated fatty acid-enriched diet increases it, while saturated fatty acids do not exert any effect; (iii) the transformation ratio is higher in diets containing starch with respect to sucrose-rich diets; (iv) caloric restriction drastically suppresses biosynthesis [18,21,22,23,24,25,26][18][21][22][23][24][25][26].
Among hormones, estrogens, glucorticoids and thyroxine are the best characterized modulators of the KP. Estrogens enhance TDO activity; enzyme activity is triplicated in women who are pregnant or are taking oral contraceptives [27,28][27][28]. Glucocorticoids stimulate de novo synthesis, by inducing TDO via a mechanism potentiated by glucagon and inhibited by insulin and adrenaline [18,29,30][18][29][30]. The effects of thyroxine on TDO activity are still controversial, as some studies suggested a positive action, while others did not observe any effect [31,32,33,34][31][32][33][34].
Due to individual differences, it has been estimated that, in human healthy individuals, Trp is converted to niacin with an average conversion efficiency of 60:1 [35]. Therefore, niacin intakes are expressed as niacin equivalents (NE; 1 mg NE = 1 mg niacin or 60 mg Trp): Recommended Dietary Allowance for adults is 16 mg NE/day for men and 14 mg NE/day for women, with a Tolerable Upper Intake Level of 35 mg/day, based on flushing as the critical adverse effect [36].
3. Pharmacological Effects of Niacin
When supplemented at physiological amounts, nicotinic acid (15–20 mg/day) and nicotinammide (300 mg/day) are effective in treating traditional pellagra [77,78][37][38]; nonetheless, at higher concentrations, they display separate additional pharmacological activities, ranging from anti-dyslipidemic to anti-inflammatory action. The first evidence of lipid-altering effects of niacin dates back to 1955, when Altschul and co-workers reported the ability of 3000 mg/day nicotinic acid (but not nicotinamide) to reduce serum cholesterol in humans [79][39]. An every growing body of experimental data points to beneficial effects of nicotinic acid as an anti-hyperlipidemic agent. It is now well established that nicotinic acid efficaciously: (i) inhibits free fatty acid mobilization and lipolysis; (ii) reduces hepatic triglyceride synthesis and very low density lipoprotein (VLDL) secretion; (iii) inhibits VLDL conversion into low density lipoprotein (LDL); (iv) increases serum high-density-lipoprotein (HDL) levels; (v) triggers LDL conversion from small, dense particles to large, low density particles, (vi) reduces serum lipoprotein concentrations; and (vii) increases apolipoprotein A1 [80,81][40][41].
To date, the underlying mechanisms are still speculative; in particular, nicotinic acid (at levels higher than those achieved with diet) has been reported to bind to and activate GPR109A and GPR109B, two G0/Gi-coupled membrane receptors highly expressed in adipose tissue; nonetheless, these receptors are absent, or present only at low levels, in the liver [82][42]. Therefore, it is conceivable that nicotinic acid might exert its lowering-lipid effects through receptor-independent and -dependent mechanisms.
Due to the above mentioned positive effects, in 2008, nicotinic acid was commercially available as Trevaclyn®, Tredaptive® or Pelzont®, at the dose of 1.0 g (in combination with laropipram, an anti-flushing agent); this prescription product has been used to treat mixed dyslipidemic and/or primary hypercholesterolemic adults receiving statins [83][43]. However, results from the Atherothrombosis Intervention in Metabolic Syndrome with Low HDL/High Triglycerides: Impact on Global Health Outcomes (AIM-HIGH) trial [84][44], together with the Heart Protection Study 2-Treatment of HDL to Reduce the Incidence of Vascular Events (HPS2-THRIVE) trial [85[45][46],86], reported no clinical benefits (i.e., reduced risk of heart attack and stroke) from the long-lasting usage of niacin. A lack of efficacy, together with the onset of recurrent serious side effects (gastrointestinal, musculoskeletal, and skin-related), has led to drug withdrawal from the EU market.
In vitro and in vivo studies have also demonstrated that nicotinic acid (or activation of its molecular targets) exerts significant anti-inflammatory, anti-oxidant and anti-apoptotic activities in a variety of cells and tissues [87][47], thus being potentially beneficial for the management of several pathological conditions, including type-2 diabetes [88[48][49],89], obesity [90[50][51],91], atherosclerosis [92][52], kidney and lung injury [93[53][54][55],94,95], and hyperalgesia [96][56].
Also nicotinamide at high doses can exert specific pharmacological activities, particularly those related to cancer management. Indeed, several experimental and clinical studies have shown the ability of nicotinamide to sensitize tumors to radiation or chemotherapy [97,98,99,100][57][58][59][60]. Such an activity depends on activation of poly(ADP-ribose)-dependent apoptosis cascade, as well as on inhibition of myosin light chain kinase that, in turn, enhances microvascular flow, thus improving drug delivery and tumor oxygenation [97,98,99,100][57][58][59][60].
4. Niacin in the Central Nervous System
Besides dermatitis and diarrhea, niacin/tryptophan deficit symptoms also include several nervous system pathologies, such as dementia and depression, as well as other symptoms resembling those observed in neurodegenerative diseases. This evidence, together with accumulating in vitro and in vivo studies, has underlined the importance of niacin (particularly of nicotinamide) in growth and maintenance of the central nervous system (CNS) [101,102][61][62].
Nicotinamide biosynthesis actively occurs in the mammalian brain, which contains nanomolar-low micromolar concentrations of nicotinamide precursors derived from the KP [103,104,105][63][64][65]. Among them, quinolinic acid (unevenly present in different brain regions and, unlike nicotinamide, unable to cross the blood-brain barrier) displays evident neuroactivity [106][66]: it acts as a N-methyl-d-aspartate (NMDA) receptor agonist, thus causing excitotoxic neuronal lesions and oxidative stress [107][67]. In addition, quinolinic acid concentrations in the brain (particularly in the cortex) positively correlate with age, thus contributing to neuron synapsis dropout occurring during aging [108][68]. Finally, neuroinflammation, neurodegeneration and mood disturbs are accompanied by increased quinolinic acid levels in plasma and/or cerebrospinal fluid [10,109,110][10][69][70].
Among KP enzymes, TDO activity is rather low in a healthy human healthy brain [111][71], where it controls neurogenesis with implications in pre- and post-natal development, as well as in anxiety-related behavior [112][72]. TDO activity is enhanced under pathological conditions: high activity, indeed, has been found in neurodegenerative diseases and during tumor progression [113,114][73][74]. Also indolamine-pyrrole 2-3 dioxygenase (IDO) is expressed in the brain and its activity is increased upon pathological conditions, especially in depression, aging and neuroinflammatory diseases [115,116,117][75][76][77].
Like other vitamins (ascorbic acid, calcitriol and retinoic acid) [118,119,120,121[78][79][80][81][82],122], nicotinamide affects neurogenesis by accelerating differentiation of embryonic stem cells or neural progenitors into post-mitotic neurons [123,124][83][84]. In vitro vitamin supplementation promotes progression of undifferentiated stem cells to neural progenitors, which further mature into efficient GABAergic neurons; the pro-inducing action is time-dependent as the effects are more pronounced when the vitamin is early received early (day 0) [124][84]. Accordingly, decreased activity of NNMT (and, therefore, low levels of its metabolic product, N1-methylnicotinamide) is required for regulating pluripotency in stem cells: accumulation of NNMT’s substrates SAM and nicotinamide, indeed, promotes naïve to primed stem cell transition, by making SAM available for histone methylation and regulation of epigenetic events that control the metabolic changes occurring in early human development [125][85].
Beside the pro-differentiating action, nicotinamide also promotes neuronal survival, especially during oxidative stress conditions, and this effect is achieved via multiple mechanisms, including: (i) prevention of cytochrome c release and caspase 3- and 9-like activities, (ii) inhibition of caspase-3-mediated degradation of forkhead transcription factor (FOXO3a) and (iii) maintenance of protein kinase B (Akt)-dependent phosphorylation of FOXO3a [126][86].
CNS vascular integrity also positively correlates with NAD levels in brain, where a fine-tuned control of its metabolism occurs. As an example, heterozygous deletion of nicotinamide phosphoribosyltransferase (NAMPT) in the brain exacerbates focal ischemic stroke-induced neuronal death and brain damage [127][87], while its selective knock down in projection neurons of adult mice leads to motor dysfunction, neurodegeneration and death [128][88].
Finally, alterations of NAD metabolism are key features of Wallerian degeneration, a process occurring in crushed nerve fibers and leading to degeneration of the axon distal to the injury, representing an early event of age-related neurodegenerative disorders, as well as of chemotherapy-induced peripheral neuropathy [129][89]. By inducing intra-axonal Ca2+ increase through a pathway requiring the action of the pro-axon death protein SARM1, accumulation of nicotinamide mononucleotide is, indeed, responsible for loss of axonal integrity [130][90]. The pro-degenerative action of nicotinamide mononucleotide has also been documented during vincristine-induced degeneration in dorsal root ganglion axons [131][91]. Accordingly, increased activity of nicotinamide/nicotinic acid-mononucleotide-adenylyltransferase (NMNAT) 1–3 protects axons from degeneration, by either limiting nicotinamide mononucleotide levels or activating SIRT1 [132,133][92][93].
References
- Spies, T.D.; Bean, W.B.; Stone, R.E. The Treatment of Subclinical and Classic Pellagra Use of Nicotinic Acid, Nicotinic Acid Amide and Sodium Nicotinate, with Special Reference to the Vasodilator Action and the Effect on Mental Symptoms. JAMA 1938, 111, 584–592.
- Magni, G.; Amici, A.; Emanuelli, M.; Orsomando, G.; Raffaelli, N.; Ruggieri, S. Enzymology of NAD+ Homeostasis in Man. Cell. Mol. Life Sci. 2004, 61, 19–34.
- Goodman, R.P.; Calvo, S.E.; Mootha, V.K. Spatiotemporal Compartmentalization of Hepatic NADH and NADPH Metabolism. J. Biol. Chem. 2018, 293, 7508–7516.
- Kirkland, J.B. Niacin Status, NAD Distribution and ADP-Ribose Metabolism. Curr. Pharm. Des. 2009, 15, 3–11.
- Nikiforov, A.; Kulikova, V.; Ziegler, M. The Human NAD Metabolome: Functions, Metabolism and Compartmentalization. Crit. Rev. Biochem. Mol. Biol. 2015, 50, 284–297.
- Anderson, K.A.; Madsen, A.S.; Olsen, C.A.; Hirschey, M.D. Metabolic Control by Sirtuins and Other Enzymes that Sense NAD(+), NADH, or Their Ratio. Biochim. Biophys. Acta Bioenerg. 2017, 1858, 991–998.
- Mutafova-Yambolieva, V.N.; Hwang, S.J.; Hao, X.; Chen, H.; Zhu, M.X.; Wood, J.D.; Ward, S.M.; Sanders, K.M. Beta-Nicotinamide Adenine Dinucleotide is an Inhibitory Neurotransmitter in Visceral Smooth Muscle. Proc. Natl. Acad. Sci. USA 2007, 104, 16359–16364.
- Moreschi, I.; Bruzzone, S.; Nicholas, R.A.; Fruscione, F.; Sturla, L.; Benvenuto, F.; Usai, C.; Meis, S.; Kassack, M.U.; Zocchi, E.; De Flora, A. Extracellular NAD+ Is an Agonist of the Human P2Y11 Purinergic Receptor in Human Granulocytes. J. Biol. Chem. 2008, 281, 31419–31429.
- Klein, C.; Grahnert, A.; Abdelrahman, A.; Muller, C.E.; Hauschildt, S. Extracellular NAD(+) Induces a Rise in (i) in Activated Human Monocytes via Engagement of P2Y(1) and P2Y(11) Receptors. Cell Calcium 2009, 46, 263–272.
- Schwarcz, R.; Stone, T.W. The Kynurenine Pathway and the Brain: Challenges, Controversies and Promises. Neuropharmacology 2016, 112, 237–247.
- Prousky, J.; Millman, C.; Kirkland, J. Pharmacologic Use of Niacin. J. Evid.-Based Complement. Altern. Med. 2011, 16, 91–101.
- Bahn, A.; Hagos, Y.; Reuter, S.; Balen, D.; Brzica, H.; Krick, W.; Burckhardt, B.C.; Sabolic, I.; Burckhardt, G. Identification of a New Urate and High Affinity Nicotinate Transporter, hOAT10 (SLC22A13). J. Biol. Chem. 2008, 283, 16332–16341.
- Kumar, J.S.; Subramanian, V.S.; Kapadia, R.; Kashyap, M.L.; Said, H.M. Mammalian Colonocytes Possess a Carrier-Mediated Mechanism for Uptake of Vitamin B3 (niacin): Studies Utilizing Human and Mouse Colonic Preparations. Am. J. Physiol. Gastrointest. Liver Physiol. 2013, 305, 207–213.
- Gopal, E.; Miyauchi, S.; Martin, P.M.; Ananth, S.; Roon, P.; Smith, S.B.; Ganapathy, V. Transport of Nicotinate and Structurally Related Compounds by Human SMCT1 (SLC5A8) and Its Relevance to Drug Transport in the Mammalian Intestinal Tract. Pharm. Res. 2007, 24, 575–584.
- Gopal, E.; Fei, Y.J.; Miyauchi, S.; Zhuang, L.; Prasad, P.D.; Ganapathy, V. Sodium-Coupled and Electrogenic Transport of B-Complex Vitamin Nicotinic Acid by slc5a8, a Member of the Na/glucose Co-transporter Gene Family. Biochem. J. 2005, 388, 309–316.
- Spector, R. Niacinamide Transport Through the Blood-Brain Barrier. Neurochem. Res. 1987, 12, 27–31.
- Badawy, A.A. Tryptophan Metabolism in Alcoholism. Nutr. Res. Rev. 2002, 15, 123–152.
- Badawy, A.A. Kynurenine Pathway of Tryptophan Metabolism: Regulatory and Functional Aspects. Int. J. Tryptophan Res. 2017, 10.
- Müller, F. Flavin-Dependent Hydroxylases. Biochem. Soc. Trans. 1985, 13, 443–447.
- Shibata, K.; Kobayashi, R.; Fukuwatari, T. Vitamin B1 Deficiency Inhibits the Increased Conversion of Tryptophan to Nicotinamide in Severe Food-Restricted Rats. Biosci. Biotechnol. Biochem. 2015, 79, 103–108.
- Shibata, K.; Onodera, M. Comparison of Tryptophan-Niacin Conversion in Rats Fed with a Nicotinic Acid-Free Diet Containing Egg White, Egg White Proteolysate, or Mixtures of Amino Acids. Agric. Biol. Chem. 1991, 55, 1291–1298.
- Shibata, K.; Nomamoto, R.; Iwai, K. Effect of Dietary Protein levels on the Urinary Excretion of Nicotinamide and Its Metabolites in Rats. Agric. Biol. Chem. 1988, 53, 1765–1769.
- Shibata, K. Nutritional Factors that Regulate on the Conversion of L-Tryptophan to Niacin. Adv. Exp. Med. Biol. 1999, 467, 711–716.
- Shibata, K. Organ Co-Relationship in Tryptophan Metabolism and Factors That Govern the Biosynthesis of Nicotinamide from Tryptophan. J. Nutr. Sci. Vitaminol. 2018, 64, 90–98.
- Shibata, K.; Nakata, C.; Fukuwatari, T. Moderate Food Restriction Suppresses the Conversion of L-tryptophan to Nicotinamide in Weaning Rats. Biosci. Biotechnol. Biochem. 2014, 78, 478–481.
- Badawy, A.A. Pellagra and Alcoholism: A Biochemical Perspective. Alcohol Alcohol. 2014, 49, 238–250.
- Bender, D.A. Inhibition in Vitro of the Enzymes of the Oxidative Pathway of Tryptophan Metabolism and of Nicotinamide Nucleotide Synthesis by Benserazide, Carbidopa and Isoniazid. Biochem. Pharmacol. 1980, 29, 707–712.
- Braidman, I.P.; Rose, D.P. The Effect of Sex Hormones on the Activity of Tryptophan Oxygenase and Other Corticosteroid-Inducible Enzymes in Rat Liver. Biochem. J. 1971, 122, 28P.
- Nakamura, T.; Shinno, H.; Ichihara, A. Insulin and Glucagon as a New Regulator System for Tryptophan Oxygenase Activity Demonstrated in Primary Cultured Rat Hepatocytes. J. Biol. Chem. 1980, 255, 7533–7535.
- Nakamura, T.; Niimi, S.; Nawa, K.; Noda, C.; Ichihara, A.; Takagi, Y.; Anai, M.; Sakaki, Y. Multihormonal Regulation of Transcription of the Tryptophan 2,3-Dioxygenase Gene in Primary Cultures of Adult Rat Hepatocytes with Special Reference to the Presence of a Transcriptional Protein Mediating the Action of Glucocorticoids. J. Biol. Chem. 1987, 262, 723–732.
- Labrie, F.; Korner, A. Effect of Glucagon, Insulin, and Thyroxine on Tyrosine Transaminase and Tryptophan Pyrrolase of Rat Liver. Arch. Biochem. Biophys. 1969, 129, 75–78.
- Chiancone, F.M. Enzyme Activities in the Tryptophan → Nicotinic Acid Path in Physiopathology. Ital. J. Biochem. 1964, 13, 1–30.
- Ku, Y.; Rogers, Q.R.; Harper, A.E. Effects of Thyroxine and Cortisol on Liver Threonine Dehydratase and Tryptophan Pyrrolase in Rats Fed a High Protein Diet. Proc. Soc. Exp. Biol. Med. 1969, 130, 556–563.
- Shibata, K.; Toda, S. Effect of Thyroxine on the Metabolism of Tryptophan to Nicotinamide in Rats. Biosci. Biotechnol. Biochem. 1994, 58, 1757–1762.
- Horwitt, M.K.; Harper, A.E.; Henderson, L.M. Niacin-Tryptophan Relationships for Evaluating Niacin Equivalents. Am. J. Clin. Nutr. 1981, 34, 423–427.
- Food and Nutrition Board. Dietary Reference Intakes for Thiamine, Riboflavin, Niacin, Vitamin B6, Folate, Vitamin B12, Panthotenic Acid, Biotin and Choline; National Academy Press: Washington, DC, USA, 1998; pp. 374–389.
- Prakash, R.; Gandotra, S.; Singh, L.K.; Das, B.; Lakra, A. Rapid Resolution of Delusional Parasitosis in Pellagra with Niacin Augmentation Therapy. Gen. Hosp. Psychiatry 2008, 30, 581–584.
- World Health Organization; United Nations High Commissions for Refugees. Pellagra and Its Prevention and Control in Major Emergencies. World Health Organization. 2000. Available online: http://www.who.int/nutrition/publications/emergencies/WHO_NHD_00.10/en/ (accessed on 1 December 2018 ).
- Altschul, R.; Hoffer, A.; Stephen, J.D. Influence of Nicotinic Acid on Serum Cholesterol in Man. Arch. Biochem. Biophys. 1955, 54, 558–559.
- Zeman, M.; Vecka, M.; Perlík, F.; Staňková, B.; Hromádka, R.; Tvrzická, E.; Širc, J.; Hrib, J.; Žák, A. Pleiotropic Effects of Niacin: Current Possibilities for Its Clinical Use. Acta Pharm. 2016, 66, 449–469.
- la Paz, S.M.; Bermudez, B.; Naranjo, M.C.; Lopez, S.; Abia, R.; Muriana, F.J. Pharmacological Effects of Niacin on Acute Hyperlipemia. Curr. Med. Chem. 2016, 23, 2826–2835.
- Offermanns, S.; Colletti, S.L.; Lovenberg, T.W.; Semple, G.; Wise, A.; IJzerman, A.P. International Union of Basic and Clinical Pharmacology. LXXXII: Nomenclature and Classification of Hydroxy-carboxylic Acid Receptors (GPR81, GPR109A, andGPR109B). Pharmacol. Rev. 2011, 63, 269–290.
- Felts, A.S. Molecule of the Month. TREDAPTIVE (Nicotinic Acid/Laropiprant): A New Lipid-Modifying Therapy for the Treatment of LDL-C, HDL-C and Triglycerides. Curr. Top. Med. Chem. 2008, 8, 1310.
- AIM-HIGH Investigators; Boden, W.E.; Probstfield, J.L.; Anderson, T.; Chaitman, B.R.; Desvignes-Nickens, P.; Koprowicz, K.; McBride, R.; Teo, K.; Weintraub, W. Niacin in Patients with Low HDL Cholesterol Levels Receiving Intensive Statin Therapy. N. Engl. J. Med. 2011, 365, 2255–2267.
- HPS2-THRIVE Collaborative Group. HPS2-THRIVE Randomized Placebo-Controlled Trial in 25 673 High-Risk Patients of ER Niacin/Laropiprant: Trial Design, Pre-Specified Muscle and Liver Outcomes, and Reasons for Stopping Studytreatment. Eur. Heart J. 2013, 34, 1279–1291.
- HPS2-THRIVE Collaborative Group; Landray, M.J.; Haynes, R.; Hopewell, J.C.; Parish, S.; Aung, T.; Tomson, J.; Wallendszus, K.; Craig, M.; Jiang, L.; et al. Effects of Extended-Release Niacin with Laropiprant in High-Risk Patients. N. Engl. J. Med. 2014, 371, 203–212.
- Graff, E.C.; Fang, H.; Wanders, D.; Judd, R.L. Anti-inflammatory Effects of the Hydroxycarboxylic Acid Receptor 2. Metabolism 2016, 65, 102–113.
- Yang, S.; Li, X.; Wang, N.; Yin, G.; Ma, S.; Fu, Y.; Wei, C.; Chen, Y.; Xu, W. GPR109A Expression in the Murine Min6 Pancreatic Beta Cell Line, and Its Relation with Glucose Metabolism and Inflammation. Ann. Clin. Lab. Sci. 2015, 45, 315–322.
- Xu, X.; Lin, S.; Chen, Y.; Li, X.; Ma, S.; Fu, Y.; Wei, C.; Wang, C.; Xu, W. The Effect of Metformin on the Expression of GPR109A, NF-κB and IL-1β in Peripheral Blood Leukocytes from Patients with Type 2 Diabetes Mellitus. Ann. Clin. Lab. Sci. 2017, 47, 556–562.
- Heemskerk, M.M.; Dharuri, H.K.; van den Berg, S.A.; Jónasdóttir, H.S.; Kloos, D.P.; Giera, M.; van Dijk, K.W.; van Harmelen, V. Prolonged Niacin Treatment Leads to Increased Adipose Tissue PUFA Synthesis and Anti-Inflammatory Lipid and Oxylipin Plasmaprofile. J. Lipid Res. 2014, 55, 2532–2540.
- Wanders, D.; Graff, E.C.; White, B.D.; Judd, R.L. Niacin Increases Adiponectin and Decreases Adipose Tissue Inflammation in High Fat Diet-Fed Mice. PLoS ONE 2013, 8, 71285.
- Su, G.; Sun, G.; Liu, H.; Shu, L.; Zhang, J.; Guo, L.; Huang, C.; Xu, J. Niacin Suppresses Progression of Atherosclerosis by Inhibiting Vascular Inflammation and Apoptosis of Vascular Smooth Muscle Cells. Med. Sci. Monit. 2015, 21, 4081–4089.
- Zhou, E.; Li, Y.; Yao, M.; Wei, Z.; Fu, Y.; Yang, Z. Niacin Attenuates the Production of Pro-Inflammatory Cytokines in LPS-Induced Mouse Alveolar Macrophages by HCA2 Dependent Mechanisms. Int. Immunopharmacol. 2014, 23, 121–126.
- Cho, K.H.; Kim, H.J.; Rodriguez-Iturbe, B.; Vaziri, N.D. Niacin ameliorates oxidative stress, inflammation, proteinuria, and hypertension in rats with chronic renal failure. Am. J. Physiol. Ren. Physiol. 2009, 297, 106–113.
- Jeong, K.Y.; Suh, G.J.; Kwon, W.Y.; Kim, K.S.; Jung, Y.S.; Kye, Y.C. The Therapeutic Effect and Mechanism of Niacin on Acute Lung Injury in a Rat Model of Hemorrhagic Shock: Down-Regulation of the Reactive Oxygen Species-Dependent Nuclear Factor κB Pathway. J. Trauma Acute Care Surg. 2015, 79, 247–255.
- Godin, A.M.; Ferreira, W.C.; Rocha, L.T.; Ferreira, R.G.; Paiva, A.L.; Merlo, L.A.; Nascimento, E.B., Jr.; Bastos, L.F.; Coelho, M.M. Nicotinic Acid Induces Antinociceptive and Anti-Inflammatory Effects in Different Experimental Models. Pharmacol. BiochemBehav. 2012, 101, 493–498.
- Ruddock, M.W.; Hirst, D.G. Nicotinamide Relaxes Vascular Smooth Muscle by Inhibiting Myosin Light Chain Kinase-Dependent Signaling Pathways: Implications for Anticancer Efficacy. Oncol. Res. 2004, 14, 483–489.
- Ruddock, M.W.; Burns, D.M.; McKeown, S.R.; Murphy, L.; Walsh, I.K.; Keane, P.F.; Hirst, D.G. Contractile Properties of Human Renal Cell Carcinoma Recruited Arteries and Theirresponse to Nicotinamide. Radiother. Oncol. 2000, 54, 179–184.
- Agote, M.; Viaggi, M.; Kreimann, E.; Krawiec, L.; Dagrosa, M.A.; Juvenal, G.J.; Pisarev, M.A. Influence of Nicotinamide on the Radiosensitivity of Normal and Goitrous Thyroid in the Rat. Thyroid 2001, 11, 1003–1007.
- Hoskin, P.; Rojas, A.; Saunders, M. Accelerated Radiotherapy, Carbogen, and Nicotinamide (ARCON) in the Treatment of Advanced Bladder Cancer: Mature Results of a Phase II Nonrandomized Study. Int. J. Radiat. Oncol. Biol. Phys. 2009, 73, 1425–1431.
- Williams, A.; Ramsden, D. Nicotinamide: A Double Edged Sword. Parkinsonism Relat. Disord. 2005, 11, 413–420.
- Fricker, R.A.; Green, E.L.; Jenkins, S.I.; Griffin, S.M. The Influence of Nicotinamide on Health and Disease in the Central Nervous System. Int. J. Tryptophan Res. 2018, 11.
- Turski, W.A.; Nakamura, M.; Todd, W.P.; Carpenter, B.K.; Whetsell, W.O., Jr.; Schwarcz, R. Identification and Quantification of Kynurenic Acid in Human Brain Tissue. Brain Res. 1988, 454, 164–169.
- Gobaille, S.; Kemmel, V.; Brumaru, D.; Dugave, C.; Aunis, D.; Maitre, M. Xanthurenic Acid Distribution, Transport, Accumulation and Release in the Rat Brain. J. Neurochem. 2008, 105, 982–993.
- Baran, H.; Schwarcz, R. Presence of 3-hydroxyanthranilic Acid in Rat Tissues and Evidence for Its Production from Anthranilic Acid in the Brain. J. Neurochem. 1990, 55, 738–744.
- Fukui, S.; Schwarcz, R.; Rapoport, S.I.; Takada, Y.; Smith, Q.R. Blood-brain Barrier Transport of Kynurenines: Implications for Brain Synthesis and Metabolism. J. Neurochem. 1991, 56, 2007–2017.
- Foster, A.C.; Collins, J.F.; Schwarcz, R. On the Excitotoxic Properties of Quinolinic Acid, 2,3-Piperidine Dicarboxylic Acids and Structurally Related Compounds. Neuropharmacology 1983, 22, 1331–1342.
- Moroni, F.; Lombardi, G.; Carlà, V.; Moneti, G. The Excitotoxin Quinolinic Acid is Present and Unevenly Distributed in the Rat Brain. Brain Res. 1984, 19, 352–355.
- Bohár, Z.; Toldi, J.; Fülöp, F.; Vécsei, L. Changing the Face of Kynurenines and Neurotoxicity: Therapeutic Considerations. Int. J. Mol. Sci. 2015, 16, 9772–9793.
- Majewski, M.; Kozlowska, A.; Thoene, M.; Lepiarczyk, E.; Grzegorzewski, W.J. Overview of the Role of Vitamins and Minerals on the Kynurenine Pathway in Health and Disease. J. Physiol. Pharmacol. 2016, 67, 3–19.
- Dang, Y.; Dale, W.E.; Brown, O.R. Comparative Effects of Oxygen on Indoleamine 2,3-Dioxygenase and Tryptophan 2,3-Dioxygenase of the Kynurenine Pathway. Free Radic. Biol. Med. 2000, 28, 615–624.
- Kanai, M.; Nakamura, T.; Funakoshi, H. Identification and Characterization of Novel Variants of the Tryptophan 2,3-Dioxygenase Gene: Differential Regulation in the Mouse Nervous System During Development. Neurosci. Res. 2009, 64, 111–117.
- Opitz, C.A.; Litzenburger, U.M.; Sahm, F.; Ott, M.; Tritschler, I.; Trump, S.; Schumacher, T.; Jestaedt, L.; Schrenk, D.; Weller, M.; et al. An Endogenous Tumour-Promoting Ligand of the Human Aryl Hydrocarbon Receptor. Nature 2011, 478, 197–203.
- Lanz, T.V.; Williams, S.K.; Stojic, A.; Iwantscheff, S.; Sonner, J.K.; Grabitz, C.; Becker, S.; Böhler, L.I.; Mohapatra, S.R.; Sahm, F.; et al. Tryptophan-2,3-Dioxygenase (TDO) Deficiency is Associated with Subclinical Neuroprotection in a Mouse Model of Multiplesclerosis. Sci. Rep. 2017, 7, 41271.
- O’Connor, J.C.; André, C.; Wang, Y.; Lawson, M.A.; Szegedi, S.S.; Lestage, J.; Castanon, N.; Kelley, K.W.; Dantzer, R. Interferon-Gamma and Tumor Necrosis Factor-Alpha Mediate the Upregulation of Indoleamine 2,3-Dioxygenase and the Induction of Depressive-Like Behavior in Mice in Response to Bacillus Calmette-Guerin. J. Neurosci. 2009, 29, 4200–4209.
- O’Connor, J.C.; Lawson, M.A.; André, C.; Briley, E.M.; Szegedi, S.S.; Lestage, J.; Castanon, N.; Herkenham, M.; Dantzer, R.; Kelley, K.W. Induction of IDO by Bacille Calmette-Guérin is Responsible for Development of Murine Depressive-Like Behavior. J. Immunol. 2009, 182, 3202–3212.
- Corona, A.W.; Norden, D.M.; Skendelas, J.P.; Huang, Y.; O’Connor, J.C.; Lawson, M.; Dantzer, R.; Kelley, K.W.; Godbout, J.P. Indoleamine 2,3-Dioxygenase Inhibition Attenuates Lipopolysaccharide Induced Persistent Microglial Activation and Depressive-Like Complications in Fractalkine Receptor (CX(3)CR1)-Deficient Mice. Brain Behav. Immun. 2013, 31, 134–142.
- Rharass, T.; Lantow, M.; Gbankoto, A.; Weiss, D.G.; Panáková, D.; Lucas, S. Ascorbic Acid Alters Cell Fate Commitment of Human Neural Progenitors in a WNT/β-Catenin/ROS Signaling Dependent Manner. J. Biomed. Sci. 2017, 16, 78.
- Cataldi, S.; Arcuri, C.; Hunot, S.; Mecca, C.; Codini, M.; Laurenti, M.E.; Ferri, I.; Loreti, E.; Garcia-Gil, M.; Traina, G.; et al. Effect of Vitamin D in HN9.10e Embryonic Hippocampal Cells and in Hippocampus from MPTP-Induced Parkinson’s Disease Mouse Model. Front. Cell. Neurosci. 2018, 12, 31.
- Cui, X.; Gooch, H.; Petty, A.; McGrath, J.J.; Eyles, D. Vitamin D and the Brain: Genomicand Non-Genomic Actions. Mol. Cell. Endocrinol. 2017, 453, 131–143.
- Haushalter, C.; Asselin, L.; Fraulob, V.; Dollé, P.; Rhinn, M. Retinoic Acid Controls Early Neurogenesis in the Developing Mouse Cerebral Cortex. Dev. Biol. 2017, 430, 129–141.
- Lin, Y.; Lin, Y.; Lin, H.; Chen, Y.; Wang, H.; Shi, J. Application of Propyl Gallate Alleviates Pericarp Browning in Harvested Longan Fruit by Modulating Metabolisms of Respiration and Energy. Food Chem. 2018, 240, 863–869.
- Griffin, S.M.; Pickard, M.R.; Orme, R.P.; Hawkins, C.P.; Fricker, R.A. Nicotinamide Promotes Neuronal Differentiation of Mouse Embryonic Stem Cells in Vitro. Neuroreport 2013, 24, 1041–1046.
- Griffin, S.M.; Pickard, M.R.; Orme, R.P.; Hawkins, C.P.; Williams, A.C.; Fricker, R.A. Nicotinamide Alone Accelerates the Conversion of Mouse Embryonic Stem Cells into Mature Neuronal Populations. PLoS ONE 2017, 12, e0183358.
- Sperber, H.; Mathieu, J.; Wang, Y.; Ferreccio, A.; Hesson, J.; Xu, Z.; Fischer, K.A.; Devi, A.; Detraux, D.; Gu, H.; et al. The Metabolome Regulates the Epigenetic Landscape During Naive-to-Primed Human Embryonic Stem Cell Transition. Nat. Cell Biol. 2015, 17, 1523–1535.
- Chong, Z.Z.; Lin, S.H.; Maiese, K. The NAD+ Precursor Nicotinamide Governs Neuronal Survival During Oxidative Stress Through Protein Kinase B Coupled to FOXO3a and Mitochondrial Membrane Potential. J. Cereb. Blood Flow Metab. 2004, 24, 728–743.
- Zhang, W.; Xie, Y.; Wang, T.; Bi, J.; Li, H.; Zhang, L.Q.; Ye, S.Q.; Ding, S. Neuronal Protective Role of PBEF in a Mouse Model of Cerebral Ischemia. J. Cereb. Blood Flow Metab. 2010, 30, 1962–1971.
- Wang, X.; Zhang, Q.; Bao, R.; Zhang, N.; Wang, Y.; Polo-Parada, L.; Tarim, A.; Alemifar, A.; Han, X.; Wilkins, H.M.; et al. Deletion of Nampt in Projection Neurons of Adult Mice Leads to Motor Dysfunction, Neurodegeneration, and Death. Cell Rep. 2017, 20, 2184–2200.
- Conforti, L.; Gilley, J.; Coleman, M.P. Wallerian Degeneration: An Emerging Axon Death Pathway Linking Injury and Disease. Nat. Rev. Neurosci. 2014, 15, 394–409.
- Loreto, A.; Di Stefano, M.; Gering, M.; Conforti, L. Wallerian Degeneration Is Executed by an NMN-SARM1-Dependent Late Ca2+ Influx butOnly Modestly Influenced by Mitochondria. Cell Rep. 2015, 13, 2539–2552.
- Liu, H.W.; Smith, C.B.; Schmidt, M.S.; Cambronne, X.A.; Cohen, M.S.; Migaud, M.E.; Brenner, C.; Goodman, R.H. Pharmacological Bypass of NAD(+) Salvage Pathway Protects Neurons from Chemotherapy-induced Degeneration. Proc. Natl. Acad. Sci. USA 2018, 115, 10654–10659.
- Araki, T.; Sasaki, Y.; Milbrandt, J. Increased Nuclear NAD Biosynthesis and SIRT1 Activation Prevent Axonal Degeneration. Science 2004, 305, 1010–1013.
- Gilley, J.; Coleman, M.P. Endogenous Nmnat2 Is an Essential Survivalfactor for Maintenance of Healthy Axons. PLoS Biol. 2010, 8, e1000300.
More