Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Conner Chen and Version 1 by Alexander E. Berezin.
Extracellular vesicles (EVs) are defined as a heterogenic group of lipid bilayer vesicular structures with a size in the range of 30–4000 nm that are released by all types of cultured cells. EVs derived from platelets, mononuclears, endothelial cells, and adipose tissue cells significantly increase in several cardiovascular diseases, including in atrial fibrillation (AF). EVs are engaged in cell-to-cell cooperation, endothelium integrity, inflammation, and immune response and are a cargo for several active molecules, such as regulatory peptides, receptors, growth factors, hormones, and lipids. Being transductors of the intercellular communication, EVs regulate angiogenesis, neovascularization, coagulation, and maintain tissue reparation.
- extracellular vesicles
- atrial fibrillation
- thrombogenicity
- biomarkers
- prediction
1. Introduction
Atrial fibrillation (AF) is the most common form of cardiac arrhythmia amongst older people and patients with cardiovascular (CV) diseases (CVD) and continues to demonstrate steady growth in the general population [1]. The prevalence of AF is increasing at epidemic proportions in both developed and developing countries, regardless of the presence of conventional CV risk factors [2,3][2][3]. Indeed, the current prevalence of AF in the European Union is 7.3%, and it will increase to 89% by 2060 [1,3][1][3]. Moreover, at least 65% of senior citizens in the European Union will have AF in 2060, and paroxysmal, persistent, and permanent forms of AF are projected to be diagnosed in approximately 5,989,000, approximately 2,833,000, and approximately 5,579,000 of older people, respectively [3]. Electroanatomic and adverse cardiac remodeling resulting in natural CVD evolution and AF persistence is considered to be a substrate for the development of cardiac dysfunction, heart failure (HF) occurrence, and thromboembolic complications, which sufficiently reduce life span duration and quality of life in patients affected by this condition [4]. In addition, AF is independently associated with both high rates of morbidity, and mortality is a common cause of premature disability in CVD patients [5]. Overall, the risk of thromboembolism in these patients is not just associated with AF, but it also varies widely depending on coexisting comorbidities (HF, chronic kidney disease, chronic obstructive pulmonary disease, diabetes mellitus, obesity, and cardiomyopathy), low physical activity, patient age and gender, and methods of cardioversion and anticoagulation [6,7,8][6][7][8]. Despite the numerous benefits of CVD prevention therapy and its wide implementation in routine practice, there remains an unacceptably high risk of potentially devastating complications associated with catheter pulmonary vein isolation (PVI), such as stroke/transient ischemic attack and systemic thromboembolism during persistent AF or the occurrence of its permanent form [9]. The number of one-year recurrent AF episodes has remained high (20%) [10], whereas single-procedural 1-year and 5-year arrhythmia-free survival is 66% and 44%, respectively [11,12][11][12]. However, the prevention of thromboembolic complications remains the focus of pragmatic strategy development for AF therapy [13,14][13][14].
The extracellular vesicles (EVs) are vesicular structures that are secreted from numerous cells and supply several biologically active molecules (growth factors, active peptides, regulatory proteins, pro-inflammatory cytokines, micro-RNAs (miR)) that are involved in cell-to-cell communication, including in the modulation of tissue repair, inflammation, angiogenesis/neovascularization, immune response, extracellular matrix accumulation, and vascular integrity enhancement [15,16,17][15][16][17]. Moreover, EVs that originate from the activated or apoptotic cells exert variable effects that are dependent on a spectrum of encapsulated cytokines, proteome, lipidome, and miRs and have an epigenetic impact on target cells [16]. There is strong evidence of the fact that EVs that are derived from a large spectrum of circulating blood cells, including platelets, mononuclear cells/leucocytes, endothelial cells, and even adipose tissue cells and antigen-presenting cells, are a cargo for pro-coagulant phospholipids, mainly phosphatidylserine, active molecules, non-coding RNAs, and other components, that regulate coagulation cascade and play a crucial role in the incidence of AF-related thromboembolism [15,16,17][15][16][17].
2. Extracellular Vesicles: Definition, Nomenclature, Biological Function
According to the International Society on Extracellular Vesicles (ISEV), EVs are defined as a heterogenic group of lipid bilayer vesicular structures with a size in the range of 30–4000 nm that are released by all types of cultured cells and are found in abundance in body fluids (blood, lymph, saliva, urine, bile, synovial fluid, cerebrospinal fluid) [18]. EVs consist of three subpopulations: exosomes, microvesicles (MVs), and apoptotic bodies (ABs), that can be distinguished from each other based on their size, immune phenotypes, origin, biogenesis, and mechanism of release and component delivery [19,20,21][19][20][21]. Table 1 contains the main characteristics of EVs. Although a universal definition of EVs subpopulations remain elusive, the ISEV recommends not using other criteria apart from the size of the EVs to classify them because there is overlap among the different phenotypes of these vesicular structures [22]. Biological pathways of generation, shedding, and the release of EVs exert remarkable diversity. For instance, exosomes appear following endocytosis from endosomes, whereas MVs are synthesized after blebbing from the plasma membrane and being released in biological fluid and contain all of the antigens that are widely expressed on the surface of the mother cells [23]. In addition, MVs and exosomes are not only produced by cellular activation, but they are also produced via pro-inflammatory stimulation, shear stress, and the influence of pro-thrombotic and pro-apoptotic substances. ABs occur as the result of the shrinkage and blebbing of apoptotic cells, but there is alternative pathway that relates to lysosome vesicle secretion and secretory autophagy. Therefore, mechanical activation and hemolysis, which are common features in patients with CVD (valve stenosis, prosthetic valves, AF, HF) as well as non-CVD (infections, sepsis, and eclampsia), are strongly associated with apoptotic cell breakdown [24,25,26,27][24][25][26][27]. In fact, an evaluation of the EVs secretome demonstrated that different sub-populations of EVs had either overlapping protein components or coincidentally similar protein arrangements, and this was a consequence of the type of stimulation that caused the EVs to release and the type of the mother cells [28].
Table 1.
Characteristics of EVs.
| Characteristics | Exosomes | Microvesicles | Apoptotic Bodies |
|---|---|---|---|
| Diameter, nm | 30–150 | 100–1000 | 500–4000 |
| Sedimentation, g | 100,000 | 20,000 | 16,000 |
| Pathway for biogenesis | Endocytosis from endosomes and exocytosis of late endosomes/MVBs | Blebbing from plasma membranes | Shrinkage and blebbing of apoptotic cells |
| Unconventional secretion pathway | Cellular activation | Early apoptosis | Lysosome vesicle secretion and secretory autophagy |
| Delivery contents | Alix, chaperones, Rab proteins, Rab GTPases, SNAREs, lipid rafts, proteins (flotillin), myokines, inflammatory cytokines, growth factors, miRs. | Arachidonic acid, cytokines, chemokine RANTES/CCL5, P-selectin, lipids, signaling proteins, miRNA, and microRNA, membrane-anchored receptors (PPARγ) and adhesion molecules | Organelles and/or nuclear content including chromatin, DNA, miRNAs, microRNAs, histones, oncogenes. |
| Membrane-specific antigens | Tetraspanins (CD9, CD81, CD63), TSG101, | Integrins, selectins, membrane proteins of parental cells | Annexin-V(+) |
Abbreviations: Alix, ALG2 interacting protein X; TSG101, tumor susceptibility gene 101; SNAREs, soluble N-ethylmaleimide-sensitive factor attachment protein receptors; PPARγ, peroxisome proliferator-activated receptor γ; MVBs, multivesicular bodies; RANTES, Regulated upon Activation, Normal T Cell-Expressed and Presumably Secreted; miRs, micro-RNAs; (+), positive.
The exact circulation levels of the different EVs subpopulations in healthy individuals and in patients with CVD and non-CVD is unclear due to a lack of strong evidence regarding their concentrations due to high variability in the sensitivity and specificity of the detection methods used or the co-detection of contaminants [29]. However, it is widely agreed upon that the majority of circulating EVs are of a platelet-derived vesicular structure, whereas neutrophil-derived, mononuclear-derived, endothelial cell-derived, and red blood cell-derived vesicles have been found in by far lower concentrations when compared to platelet-derived ones [30].
Nowadays, EVs are considered to be powerful mediator of cell-to-cell communication through the delivery of cargo proteins (adenosine diphosphate-ribosylation factor 6, Ras-related protein 22a, vesicle-soluble NSF attachment protein receptor, vesicle-associated membrane protein 3, T-cell internal antigen 1, argonaute-2, lipids, mRNA, miRNA long noncoding RNA, and occasionally genomic DNA) [31]. In addition, EVs directly transfer functional receptors, such as CD41, CD61, CD62, CXCR4, PAR-1, and PPRγ, from parental cells to recipient cells, contributing an axis for the regulation of their proliferation, differentiation, and activation. A large spectrum of miRNAs was observed in the EVs that were isolated from human parental cells. More often than not, the miRNAs that are incorporated into EVs act as negative regulators for several biological processes, including tissue reparation, vascular integrity, angiogenesis, neovascularization, immune response, and inflammation. Indeed, miR126-3p is responsible for the suppression of the expression of different inflammatory genes in the macrophages/mononuclears and thereby mediates cytokine-induced thrombogenicity and NETosis [32]. Platelet-derived EVs enriched in miR 21, miR-223, and miR-339 seem to be a modulator of expression of the platelet-derived growth factor receptor-beta in smooth muscle cells, playing a crucial role in plaque development and vascular remodeling [33]. Figure 1 illustrates the role of EVs’ secretome depending on the origin of the vesicles.
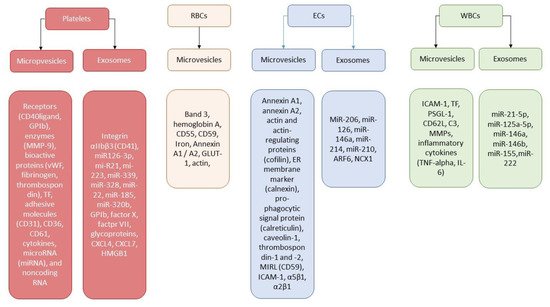
Figure 1. Secretome of EVs originated from different cells. Abbreviations: ARF6, ADP ribosylation factor 6; ECs, endothelial cells; RBCs, red blood cells; ER, endoplasmic reticulum; ICAM-1, intracellular adhesion molecule 1; CD62L, l-selectin; MIRL, membrane inhibitor of reactive lysis; MMPs, metalloproteinases; TNF-alpha, tumor necrosis factor-alpha; TF, tissue factor; vWF; Von Willebrand factor; CXCL, C-X-C motif ligand; HMGB1, high mobility group box 1; PSGL-1, P-selectin glycoprotein ligand-1; WBCs, white blood cells.
3. Extracellular Vesicles and Thrombogenicity/Thrombosis
It has been suggested that cell-derived EVs that are enriched in lipids (phosphatidylserine, arachidonic acids), secretory phospholipase A2, factors of coagulation (factor X, factor VII, tissue factor), chromatin, and DNAs/RNAs might have 50- to 100-fold higher specific pro-coagulant activity than activated platelets [30,34][30][34]. The mechanisms by which these EVs exert their pro-coagulative abilities are considered to be quite complex (Figure 2).
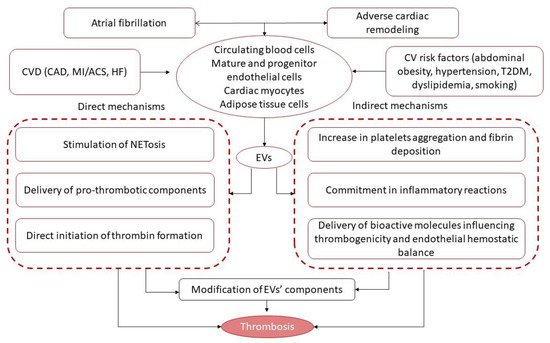
Figure 2. Pathogenetic pathways underlying EV-related thrombosis formation and increase in thrombogenicity. Abbreviations: CAD, coronary artery disease; ACS, acute coronary syndrome; EVs, extracellular vesicles; NETosis, neutrophil extracellular traps; AF, atrial fibrillation; CVD, cardiovascular diseases; CV, cardiovascular; HF, heart failure; T2DM, type 2 diabetes mellitus.
First, secreted EVs may facilitate the formation of neutrophil extracellular traps (NETs), which may contribute to thrombus development and attenuate the counteracting effect of endogenous fibrinolytic systems [35,36][35][36]. Second, the presence of polyphosphate (polyP) in cell-derived EVs may promote thrombosis through a tissue factor-independent route, whereas these effects were initially described in cancer-associated thrombosis [37] and then extrapolated to others. Yet, the pro-coagulant activity to EVs that originated from mature endothelial cells was found to be up-regulated by the induction of the expression of adhesion proteins and several encapsulated pro-inflammatory cytokines, mainly IL-8 and tumor necrosis factor-alpha (TNF-alpha), on mother cells [38]. These mechanisms are rigorously regulated by the protease-activated receptor (PAR) 2 signaling pathway, which, in turn, mediates the rapid generation of pro-thrombotic components, such as inactive tissue factor/factor VII and integrin α5 β1 into the EVs secreted by endothelial cells [39]. Finally, the internationalization of the pro-thrombotic complex consists of tissue factor–factor VIIa–factor Xa by endothelial cells and regulates tissue factor availability for release on pro-coagulant EVs [40,41][40][41]. Third, the P2Y1 and P2Y12 receptors, which play a pivotal role in platelet activation and aggregation, seem to be transported by EVs as membrane-associated structures [42]. The next thrombogenicity-stimulating pathway is direct thrombin activation by EVs enriched by chemokines (CXCL4, CXCL7) and the cytoplasmic high-mobility group box 1 protein [43]. In addition, platelet-derived and endothelial cell-derived MVs and exosomes can stimulate coagulation independently after the mechanical stimulation of the parental cells or due to hemolysis [44].
Therefore, EVs can potentiate thrombosis through several indirect mechanisms, such as through an increase in platelet aggregation and fibrin deposition following the transfer of arachidonic acid [45], the down-regulation of endothelial cyclooxygenase (COX)-2 expression and prostacyclin synthesis via thromboxane A2-related mechanisms [46], and increased endothelial cell surface thrombogenicity and altered endothelial hemostatic balance [47]. It has been found that platelet-derived MVs are able to modulate the expression of (COX)-2 and prostacyclin production in both circulating monocytes and in progenitor/mature endothelial cells through the direct activation of the PKC/p42/p44 MAPK/p38 kinase and c-Jun N-terminal kinase/Elk-1pathways [48]. Indeed, these pathways contribute to vascular homeostasis through the regulation of plasmin generation on the endothelial cell layer and is a crucial player in ensuring thrombogenicity and blood clotting control [49]. The EVs that are mainly derived from endothelial cells participate in transfer growth factors (fibroblast growth factor, transforming growth factor-beta), active proteins (including annexin A2 ligand and CD40 ligand+), and receptors, resulting in TNF-alpha and IL-6 co-stimulation, and, in turn, lead to plasmin generation and the expression of both the urokinase-type plasminogen activator (uPA) and its receptor (uPAR) on the surface of endothelial cells [50]. All of these trigger an increase in their ability to bind exogenous uPA on uPAR and to subsequently maintain plasmin formation. Moreover, this was found to be a signal messenger for endothelial cell-derived EVs to induce plasmin generation and, in turn, affect tube formation in endothelial progenitor cells, thereby ensuring their proliferative and proteolytic activities [49]. Previous studies have shown that S100A10 is a member of the S100 family of Ca2+-binding proteins and was found to be abundantly distributed in progenitor and mature endothelial cells and protected from altered vascular integrity and hypercoagulation [51,52][51][52]. Indeed, this resulted in the loss of S100A10 from the endothelial cells, which exerted significant plasmin generation suppression [51,52,53][51][52][53]. It is possible that the EVs originating from activated endothelial cells are able to be a cargo for the kringle-2 domain of tPA, which plays a crucial role in S100A10-dependent plasmin generation [54]. Thus, decreased number and lowered functional activity of endothelial cell-derived EVs are considered to be a powerful factors for increased thrombogenicity.
These mechanisms based on both the synthesis and transfer of coagulation factors by EVs, but these can also be activated via the pro-inflammatory cytokine cascade, the proteasome-dependent mechanism, and apoptosis [55]. In fact, It is prudently suggested that these mechanisms overlap with each other. For instance, pro-inflammatory cytokine IL-33 and cholesterol were found to be powerful inductors for differential tissue factor expression and stimulators for monocyte subsets as well as for the release of pro-coagulant EVs into circulation from mother cells [55,56][55][56]. Consequently, inflammatory cytokines along with other factors generated during blood coagulation, such as platelet-derived lipid mediators (lysophosphatidate, phosphatidic acid and sphingosine 1-phosphate), may contribute to the formation of a pro-thrombotic state in CVD patients as well as potentiate neovascularization and angiogenesis [57]. Considerable attention has focused on the ability of pro-coagulant lipid components, mainly phospholipids derived through EVs, to be agonists for G-protein-coupled endothelial differentiation gene receptors, which seem to be important for the morphogenesis of capillary-like structures, promoting angiogenesis and supporting blood clotting [57]. On the other hand, numerous EVs enriched in phospholipids have been derived from activated platelets, resulting in blood coagulation and were eventually noticed to be key modulators of the chemo-attractive and proteolytic activity of the endothelial cells [58,59][58][59] that linked CV factors, comorbidities, endothelial integrity, and thrombogenicity [60].
Another pathophysiologic mechanism that would explain the link between AF and thrombogenicity is the indirect influence of the EVs on thromboembolic complications through adverse cardiac remodeling. Indeed, there is a wide range of resoundingly clear scientific proof of the fact that EVs enriched in cardiac protective microRNAs, such as miR-30d, miR-17-3p, miR-155, miR-222, and miR-378, are primary secreted by cardiac myocytes following acute-phase ischemia, hypoxia, inflammation, and biochemical stress and may ameliorate apoptosis through the activation of MAP4K4 (mitogen-associate protein kinase 4), the down-regulation of tumor protein p53-inducible nuclear protein 1, and the suppression of cardiac fibroblast proliferation and activation by directly targeting integrin α5 [61,62,63][61][62][63]. On the contrary, the chronic phases of these conditions were strongly associated with lower miR-30d expression in the myocardium and also decreased the amount of circulating EVs enriched in miR-30d, miR-17-3p, and miR-222, something that that is considered to be related to adverse cardiac remodeling in animal models and humans [62,63,64][62][63][64]. All of these are consequently linked to the over-expression of the genes that are implicated in fibrosis and inflammation and might play a role in the occurrence of non-valvular AF [65]. In addition, lowered levels of circulating endothelial cell-derived EVs, mainly the MVs that are able to regulate myocardial reparation and vascular integrity through supplying active molecules, regulatory peptides, miRNAs, and growth factors, were found to be predictors of adverse cardiac remodeling and AF [66,67][66][67]. In addition, the EVs derived from adipose tissues, including epicardial fat, as well as those originating from apoptotic mononuclear cells, were found to have encapsulated certain inflammatory cytokines, such as TNF-alpha, IL-1α, IL-1β, and RANTES, and they were also determined to have a cytotoxic impact on the myocardium, promoting left ventricular hypertrophy and arhythmogenesis [68,69][68][69]. Thus, the EVs originating from cardiac myocytes and circulating blood, mainly endothelial cells and mononuclear cells, may indirectly influence the development of AF through the mediation of cardiac hypertrophy, fibrosis, and oxidative stress/inflammation and aggravating atrial remodeling.
References
- Di Carlo, A.; Bellino, L.; Consoli, D.; Mori, F.; Zaninelli, A.; Baldereschi, M.; Cattarinussi, A.; D’Alfonso, M.G.; Gradia, C.; Sgherzi, B.; et al. Prevalence of atrial fibrillation in the Italian elderly population and projections from 2020 to 2060 for Italy and the European Union: The FAI Project. EP Eur. 2019, 21, 1468–1475.
- Tedrow, U.B.; Conen, D.; Ridker, P.M.; Cook, N.R.; Koplan, B.A.; Manson, J.E.; Buring, J.E.; Albert, C.M. The long- and short-term impact of elevated body mass index on the risk of new atrial fibrillation the WHS (women’s health study). J. Am. Coll. Cardiol. 2010, 55, 2319–2327.
- Kloosterman, M.; Crijns, H.; Van Gelder, I.C. Rising prevalence of atrial fibrillation in the elderly population: New challenges of geriatric cardiology. EP Eur. 2019, 21, 1451–1453.
- Carlisle, M.A.; Fudim, M.; DeVore, A.D.; Piccini, J.P. Heart Failure and Atrial Fibrillation, Like Fire and Fury. JACC Heart Fail. 2019, 7, 447–456.
- Hua, C.; Jiang, C.; He, L.; Jia, Z.X.; Lyu, W.H.; Tang, R.B.; Sang, C.H.; Long, D.Y.; Dong, J.Z.; Ma, C.S.; et al. Causes of death and influencing factors of atrial fibrillation patients undergoing anticoagulation therapy. Zhonghua Xin Xue Guan Bing Za Zhi 2021, 49, 353–359.
- Rankin, A.J.; Rankin, S.H. Cardioverting acute atrial fibrillation and the risk of thromboembolism: Not all patients are created equal. Clin. Med. 2017, 17, 419–423.
- Proietti, M.; Laroche, C.; Drozd, M.; Vijgen, J.; Cozma, D.C.; Drozdz, J.; Maggioni, A.P.; Boriani, G.; Lip, G.Y.H.; EORP-AF Investigators. Impact of chronic obstructive pulmonary disease on prognosis in atrial fibrillation: A report from the EURObservational Research Programme Pilot Survey on Atrial Fibrillation (EORP-AF) General Registry. Am. Heart J. 2016, 181, 83–91.
- Boriani, G.; Laroche, C.; Diemberger, I.; Fantecchi, E.; Popescu, M.I.; Rasmussen, L.H.; Dan, G.-A.; Kalarus, Z.; Tavazzi, L.; Maggioni, A.P.; et al. ‘Real-world’ management and outcomes of patients with paroxysmal vs. non-paroxysmal atrial fibrillation in Europe: The EURObservational Research Programme-Atrial Fibrillation (EORP-AF) General Pilot Registry. EP Eur. 2016, 18, 648–657.
- Lip, G.Y.H.; Laroche, C.; Boriani, G.; Cimaglia, P.; Dan, G.-A.; Santini, M.; Kalarus, Z.; Rasmussen, L.H.; Popescu, M.I.; Tica, O.; et al. Sex-related differences in presentation, treatment, and outcome of patients with atrial fibrillation in Europe: A report from the Euro Observational Research Programme Pilot survey on Atrial Fibrillation. EP Eur. 2015, 17, 24–31.
- Sivasambu, B.; Balouch, M.A.; Zghaib, T.; Bajwa, R.J.; Chrispin, J.; Berger, R.D.; Ashikaga, H.; Nazarian, S.; Marine, J.E.; Calkins, H.; et al. Increased rates of atrial fibrillation recurrence following pulmonary vein isolation in overweight and obese patients. J. Cardiovasc. Electrophysiol. 2018, 29, 239–245.
- Scherr, D.; Khairy, P.; Miyazaki, S.; Aurillac-Lavignolle, V.; Pascale, P.; Wilton, S.B.; Ramoul, K.; Komatsu, Y.; Roten, L.; Jadidi, A.; et al. Five-year outcome of catheter ablation of persistent atrial fibrillation using termination of atrial fibrillation as a procedural endpoint. Circulation. Arrhythmia Electrophysiol. 2015, 8, 18–24.
- Saguner, A.M.; Maurer, T.; Wissner, E.; Santoro, F.; Lemes, C.; Mathew, S.; Sohns, C.; Heeger, C.H.; Reißmann, B.; Riedl, J.; et al. Catheter ablation of atrial fibrillation in very young adults: A 5-year follow-up study. EP Eur. 2018, 20, 58–64.
- Jame, S.; Barnes, G. Stroke and thromboembolism prevention in atrial fibrillation. Heart 2020, 106, 10–17.
- O’Neill, L.; Wielandts, J.Y.; Gillis, K.; Hilfiker, G.; Le Polain De Waroux, J.-B.; Tavernier, R.; Duytschaever, M.; Knecht, S. Catheter Ablation in Persistent AF, the Evolution towards a More Pragmatic Strategy. J. Clin. Med. 2021, 10, 4060.
- Blanch-Ruiz, M.A.; Ortega-Luna, R.; Martínez-Cuesta, M.Á.; Álvarez, Á. The Neutrophil Secretome as a Crucial Link between Inflammation and Thrombosis. Int. J. Mol. Sci. 2021, 22, 4170.
- Berezin, A.; Zulli, A.; Kerrigan, S.; Petrovic, D.; Kruzliak, P. Predictive role of circulating endothelial-derived microparticles in cardiovascular diseases. Clin. Biochem. 2015, 48, 562–568.
- De Freitas, R.; Hirata, R.; Hirata, M.H.; Aikawa, E. Circulating Extracellular Vesicles as Biomarkers and Drug Delivery Vehicles in Cardiovascular Diseases. Biomolecules 2021, 11, 388.
- Théry, C.; Witwer, K.W. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): A position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J. Extracell. Vesicles 2018, 7, 1535750.
- Van der Pol, E.; Böing, A.N.; Gool, E.L.; Nieuwland, R. Recent developments in the nomenclature, presence, isolation, detection and clinical impact of extracellular vesicles. J. Thromb. Haemost. JTH 2016, 14, 48–56.
- Nederveen, J.P.; Warnier, G.; Di Carlo, A.; Nilsson, M.I.; Tarnopolsky, M.A. Extracellular Vesicles and Exosomes: Insights from Exercise Science. Front. Physiol. 2021, 11, 604274.
- Askeland, A.; Borup, A.; Østergaard, O.; Olsen, J.V.; Lund, S.M.; Christiansen, G.; Kristensen, S.R.; Heegaard, N.; Pedersen, S. Mass-Spectrometry Based Proteome Comparison of Extracellular Vesicle Isolation Methods: Comparison of ME-kit, Size-Exclusion Chromatography, and High-Speed Centrifugation. Biomedicines 2020, 8, 246.
- Witwer, K.W.; Goberdhan, D.C.; O’Driscoll, L.; Théry, C.; Welsh, J.A.; Blenkiron, C.; Buzás, E.I.; Di Vizio, D.; Erdbrügger, U.; Falcón-Pérez, J.M.; et al. Updating MISEV: Evolving the minimal requirements for studies of extracellular vesicles. J. Extracell. Vesicles 2021, 10, e12182.
- Femminò, S.; Penna, C.; Margarita, S.; Comità, S.; Brizzi, M.F.; Pagliaro, P. Extracellular vesicles and cardiovascular system: Biomarkers and Cardioprotective Effectors. Vasc. Pharmacol. 2020, 135, 106790.
- Puhm, F.; Boilard, E.; Machlus, K.R. Platelet Extracellular Vesicles: Beyond the Blood. Arterioscler. Thromb. Vasc. Biol. 2021, 41, 87–96.
- Abels, E.R.; Breakefield, X.O. Introduction to Extracellular Vesicles: Biogenesis, RNA Cargo Selection, Content, Release, and Uptake. Cell. Mol. Neurobiol. 2016, 36, 301–312.
- Puhm, F.; Flamand, L.; Boilard, E. Platelet extracellular vesicles in COVID-19: Potential markers and makers. J. Leukoc. Biol. 2022, 111, 63–74.
- Berezin, A.E. Microparticles in Chronic Heart Failure. Adv. Clin. Chem. 2017, 81, 1–41.
- Bebelman, M.P.; Smit, M.J.; Pegtel, D.M.; Baglio, S.R. Biogenesis and function of extracellular vesicles in cancer. Pharmacol. Ther. 2018, 188, 1–11.
- Arraud, N.; Linares, R.; Tan, S.; Gounou, C.; Pasquet, J.M.; Mornet, S.; Brisson, A.R. Extracellular vesicles from blood plasma: Determination of their morphology, size, phenotype and concentration. J. Thromb. Haemost. 2014, 12, 614–627.
- Sinauridze, E.I.; Kireev, D.A.; Popenko, N.Y.; Pichugin, A.V.; Panteleev, M.A.; Krymskaya, O.V.; Ataullakhanov, F.I. Platelet microparticle membranes have 50- to 100-fold higher specific procoagulant activity than activated platelets. Thromb. Haemost. 2007, 97, 425–434.
- Simons, M.; Raposo, G. Exosomes—Vesicular carriers for intercellular communication. Curr. Opin. Cell Biol. 2009, 21, 575–581.
- Laffont, B.; Corduan, A.; Rousseau, M.; Duchez, A.C.; Lee, C.H.; Boilard, E.; Provost, P. Platelet microparticles reprogram macrophage gene expression and function. Thromb. Haemost. 2016, 115, 311–323.
- Silverman-Gavrila, R.; Silverman-Gavrila, L.; Bendeck, M.P. Cell division fidelity is altered during the vascular response to injury: Its novel role in atherosclerosis progression. Am. J. Pathol. 2013, 182, 628–639.
- Fourcade, O.; Simon, M.F.; Viode, C.; Rugani, N.; Leballe, F.; Ragab, A.; Fournie, B.; Sarda, L.; Chap, H. Secretory phospholipase A2 generates the novel lipid mediator lysophosphatidic acid in membrane microvesicles shed from activated cells. Cell 1995, 80, 919–927.
- Almeida, V.H.; Rondon, A.; Gomes, T.; Monteiro, R.Q. Novel Aspects of Extracellular Vesicles as Mediators of Cancer-Associated Thrombosis. Cells 2019, 8, 716.
- Berezin, A. Neutrophil extracellular traps: The core player in vascular complications of diabetes mellitus. Diabetes Metab. Syndr. 2019, 13, 3017–3023.
- Gasecka, A.; Böing, A.N.; Filipiak, K.J.; Nieuwland, R. Platelet extracellular vesicles as biomarkers for arterial thrombosis. Platelets 2017, 28, 228–234.
- Rothmeier, A.S.; Versteeg, H.H.; Ruf, W. Factor VIIa-induced interaction with integrin controls the release of tissue factor on extracellular vesicles from endothelial cells. J. Thromb. Haemost. 2019, 17, 627–634.
- Rothmeier, A.S.; Liu, E.; Chakrabarty, S.; Disse, J.; Mueller, B.M.; Østergaard, H.; Ruf, W. Identification of the integrin-binding site on coagulation factor VIIa required for proangiogenic PAR2 signaling. Blood 2018, 131, 674–685.
- Zifkos, K.; Dubois, C.; Schäfer, K. Extracellular Vesicles and Thrombosis: Update on the Clinical and Experimental Evidence. Int. J. Mol. Sci. 2021, 22, 9317.
- Rosas, M.; Slatter, D.A.; Obaji, S.G.; Webber, J.P.; Alvarez-Jarreta, J.; Thomas, C.P.; Aldrovandi, M.; Tyrrell, V.J.; Jenkins, P.V.; O’Donnell, V.B.; et al. The procoagulant activity of tissue factor expressed on fibroblasts is increased by tissue factor-negative extracellular vesicles. PLoS ONE 2020, 15, e0240189.
- Gąsecka, A.; Rogula, S.; Eyileten, C.; Postuła, M.; Jaguszewski, M.J.; Kochman, J.; Mazurek, T.; Nieuwland, R.; Filipiak, K.J. Role of P2Y Receptors in Platelet Extracellular Vesicle Release. Int. J. Mol. Sci. 2020, 21, 6065.
- Zarà, M.; Guidetti, G.F.; Camera, M.; Canobbio, I.; Amadio, P.; Torti, M.; Tremoli, E.; Barbieri, S.S. Biology and Role of Extracellular Vesicles (EVs) in the Pathogenesis of Thrombosis. Int. J. Mol. Sci. 2019, 20, 2840.
- Suades, R.; Padro, T.; Vilahur, G.; Badimon, L. Circulating and platelet-derived microparticles in human blood enhance thrombosis on atherosclerotic plaques. Thromb. Haemost. 2012, 108, 1208–1219.
- Barry, O.P.; Pratico, D.; Lawson, J.A.; FitzGerald, G.A. Transcellular activation of platelets and endothelial cells by bioactive lipids in platelet microparticles. J. Clin. Investig. 1997, 99, 2118–2127.
- Caughey, G.E.; Cleland, L.G.; Gamble, J.R.; James, M.J. Up-regulation of endothelial cyclooxygenase-2 and prostanoid synthesis by platelets. Role of thromboxane A2. J. Biol. Chem. 2001, 276, 37839–37845.
- Aharon, A.; Tamari, T.; Brenner, B. Monocyte-derived microparticles and exosomes induce procoagulant and apoptotic effects on endothelial cells. Thromb. Haemost. 2008, 100, 878–885.
- Barry, O.P.; Kazanietz, M.G.; Praticò, D.; FitzGerald, G.A. Arachidonic acid in platelet microparticles up-regulates cyclooxygenase-2-dependent prostaglandin formation via a protein kinase C/mitogen-activated protein kinase-dependent pathway. J. Biol. Chem. 1999, 274, 7545–7556.
- Lacroix, R.; Sabatier, F.; Mialhe, A.; Basire, A.; Pannell, R.; Borghi, H.; Robert, S.; Lamy, E.; Plawinski, L.; Camoin-Jau, L.; et al. Activation of plasminogen into plasmin at the surface of endothelial microparticles: A mechanism that modulates angiogenic properties of endothelial progenitor cells in vitro. Blood 2007, 110, 2432–2439.
- Plesner, T.; Behrendt, N.; Ploug, M. Structure, function and expression on blood and bone marrow cells of the urokinase-type plasminogen activator receptor, uPAR. Stem cells 1997, 15, 398–408.
- Kwon, M.; MacLeod, T.J.; Zhang, Y.; Waisman, D.M. S100A10, annexin A2, and annexin a2 heterotetramer as candidate plasminogen receptors. Front. Biosci. 2005, 10, 300–325.
- He, K.L.; Deora, A.B.; Xiong, H.; Ling, Q.; Weksler, B.B.; Niesvizky, R.; Hajjar, K.A. Endothelial cell annexin A2 regulates polyubiquitination and degradation of its binding partner S100A10/p11. J. Biol. Chem. 2008, 283, 19192–19200.
- Miller, V.A.; Madureira, P.A.; Kamaludin, A.A.; Komar, J.; Sharma, V.; Sahni, G.; Thelwell, C.; Longstaff, C.; Waisman, D.M. Mechanism of plasmin generation by S100A10. Thromb. Haemost. 2017, 117, 1058–1071.
- Bharadwaj, A.; Kempster, E.; Waisman, D.M. The Annexin A2/S100A10 Complex: The Mutualistic Symbiosis of Two Distinct Proteins. Biomolecules 2021, 11, 1849.
- Stojkovic, S.; Thulin, Å.; Hell, L.; Thaler, B.; Rauscher, S.; Baumgartner, J.; Gröger, M.; Ay, C.; Demyanets, S.; Neumayer, C.; et al. IL-33 stimulates the release of procoagulant microvesicles from human monocytes and differentially increases tissue factor in human monocyte subsets. Thromb. Haemost. 2017, 117, 1379–1390.
- Liu, M.L.; Reilly, M.P.; Casasanto, P.; McKenzie, S.E.; Williams, K.J. Cholesterol enrichment of human monocyte/macrophages induces surface exposure of phosphatidylserine and the release of biologically-active tissue factor-positive microvesicles. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 430–435.
- Harvey, K.; Siddiqui, R.A.; Sliva, D.; Garcia, J.G.; English, D. Serum factors involved in human microvascular endothelial cell morphogenesis. J. Lab. Clin. Med. 2002, 140, 188–198.
- English, D.; Welch, Z.; Kovala, A.T.; Harvey, K.; Volpert, O.V.; Brindley, D.N.; Garcia, J.G. Sphingosine 1-phosphate released from platelets during clotting accounts for the potent endothelial cell chemotactic activity of blood serum and provides a novel link between hemostasis and angiogenesis. FASEB J. 2000, 14, 2255–2265.
- Limaye, V. The role of sphingosine kinase and sphingosine-1-phosphate in the regulation of endothelial cell biology. Endothelium 2008, 15, 101–112.
- Berezin, A.E. Impaired Immune Phenotype of Endothelial Cell-derived Micro Particles: The Missing Link between Diabetes-related States and Risk of Cardiovascular Complications? J. Data Min. Genom. Proteom. 2016, 7, 195–197.
- Li, J.; Salvador, A.M.; Li, G.; Valkov, N.; Ziegler, O.; Yeri, A.; Yang Xiao, C.; Meechoovet, B.; Alsop, E.; Rodosthenous, R.S.; et al. Mir-30d Regulates Cardiac Remodeling by Intracellular and Paracrine Signaling. Circ. Res. 2021, 128, e1–e23.
- He, W.; Huang, H.; Xie, Q.; Wang, Z.; Fan, Y.; Kong, B.; Huang, D.; Xiao, Y. MiR-155 Knockout in Fibroblasts Improves Cardiac Remodeling by Targeting Tumor Protein p53-Inducible Nuclear Protein 1. J. Cardiovasc. Pharmacol. Ther. 2016, 21, 423–435.
- Yuan, J.; Liu, H.; Gao, W.; Zhang, L.; Ye, Y.; Yuan, L.; Ding, Z.; Wu, J.; Kang, L.; Zhang, X.; et al. MicroRNA-378 suppresses myocardial fibrosis through a paracrine mechanism at the early stage of cardiac hypertrophy following mechanical stress. Theranostics 2018, 8, 2565–2582.
- Schüttler, D.; Clauss, S.; Weckbach, L.T.; Brunner, S. Molecular Mechanisms of Cardiac Remodeling and Regeneration in Physical Exercise. Cells 2019, 8, 1128.
- Maries, L.; Marian, C.; Sosdean, R.; Goanta, F.; Sirbu, I.O.; Anghel, A. MicroRNAs—The Heart of Post-Myocardial Infarction Remodeling. Diagnostics 2021, 11, 1675.
- Wu, Q.; Wang, J.; Tan, W.; Jiang, Y.; Wang, S.; Li, Q.; Yu, X.; Tan, J.; Liu, S.; Zhang, P.; et al. Extracellular vesicles from human embryonic stem cell-derived cardiovascular progenitor cells promote cardiac infarct healing through reducing cardiomyocyte death and promoting angiogenesis. Cell Death Dis. 2020, 11, 354.
- Berezin, A.E.; Berezin, A.A. Extracellular Endothelial Cell-Derived Vesicles: Emerging Role in Cardiac and Vascular Remodeling in Heart Failure. Front. Cardiovasc. Med. 2020, 7, 47.
- Bang, C.; Batkai, S.; Dangwal, S.; Gupta, S.K.; Foinquinos, A.; Holzmann, A.; Just, A.; Remke, J.; Zimmer, K.; Zeug, A.; et al. Cardiac fibroblast-derived microRNA passenger strand-enriched exosomes mediate cardiomyocyte hypertrophy. J. Clin. Investig. 2014, 124, 2136–2146.
- Shaihov-Teper, O.; Ram, E.; Ballan, N.; Brzezinski, R.Y.; Naftali-Shani, N.; Masoud, R.; Ziv, T.; Lewis, N.; Schary, Y.; Levin-Kotler, L.P.; et al. Extracellular Vesicles from Epicardial Fat Facilitate Atrial Fibrillation. Circulation 2021, 143, 2475–2493.
More