Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 3 by Conner Chen and Version 2 by Conner Chen.
GM1 (monosialotetrahexosylganglioside) the "prototype" ganglioside, is a member of the ganglio series of gangliosides which contain one sialic acid residue. GM1 has important physiological properties and impacts neuronal plasticity and repair mechanisms, and the release of neurotrophins in the brain.
- Parkinson’s disease
- GM1
- ganglioside
- neuroprotection
1. Introduction
The numerous glycosphingolipids that occur in the nervous system and elsewhere are clearly involved in metabolic and pathological changes that accompany Parkinson’s disease [1], but one such molecule, GM1 ganglioside, has received focused attention from several workers for its prominent role in both the etiology and potential treatment of this neurodegenerative condition. GM1 is particularly abundant in neurons and is essential for their complex functioning (see below). The same may be said for ganglioside GD1a, which is identical in structure to GM1 but possesses an additional sialic acid attached to the terminal galactose of GM1 (Figure 1) [2]. That terminal sialic acid is readily removed by NEU3 neuraminidase, which is situated close to GD1a in the neuronal membrane [3]. This is why GD1a is considered to function as a reserve pool for GM1 as its primary function.
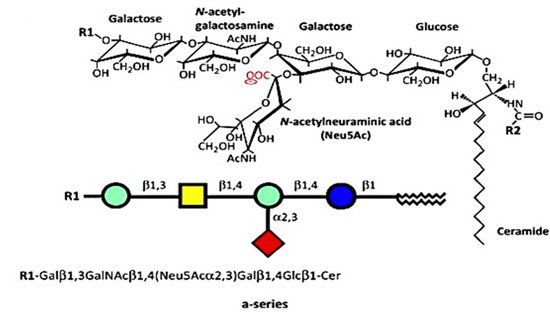
Figure 1. Structures of a-series. GM1 (GM1a), R1 = H, R2 = long-chain fatty acid (e.g., 18:0); GD1a, R1 = Neu5Acα2,3, R2 = long-chain fatty acid (e.g., 18:0); LIGA20, R1 = H, R2 = CHCl2 (image used are adapted from [4] with permission granted by Trends in biochemical sciences, Copyright 2015 Cellpress).
2. GM1 Deficiency Induces Parkinsonism in Mouse PD Model
One of the first clues suggesting that GM1 deficiency constitutes a major risk factor in PD came from the serendipitous observation of mice deficient in GM1 due to biallelic disruption of B4galnt1 (GM2 synthase-B4galnt1–/–) [5]. GM2 is the obligatory precursor to GM1. In addition to motor dysfunction, these mice showed several neuropathologies characteristic of PD, including the key feature of aggregated alpha-synuclein. Additional symptoms included the depletion of striatal dopamine (DA) and loss of DA neurons of the substantia nigra pars compacta (SNpc). Movement disorder was alleviated by L-dopa administration, and neuropathologies were alleviated by LIGA20, an analog of GM1 structurally similar to the latter (Figure 1) but is more membrane permeable and, hence, more active. GM1 itself was not effective in those initial studies due to inadequate dosages. However, other investigators showed GM1 to be therapeutically effective in PD mice under appropriate conditions [2; for review].
Study [6] was particularly revealing, in which mice with only monoallelic disruption of the same gene (B4galnt1+/−) manifested Parkinsonian symptoms closely similar to those of the above biallelic mice; such mice had only partial deficiency of GM1. In order to test the possibility that the same might apply to PD itself, we carried out immunohistochemical analysis of paraffin sections from the SNpc of PD subjects [6]. These showed substantially less GM1 in nigral DA neurons than in age-matched controls (Figure 2). Soon after that, we came into the possession of tissues from the occipital cortex of PD patients, and we measured their GM1 by the well-established method of high performance thin-layer chromatography (HPTLC) [7]. This too showed a significant deficiency of GM1, even though that part of the brain is not intimately involved in PD pathology. These findings prompted us to examine additional tissues to observe how widespread GM1 deficiency actually is in PD.
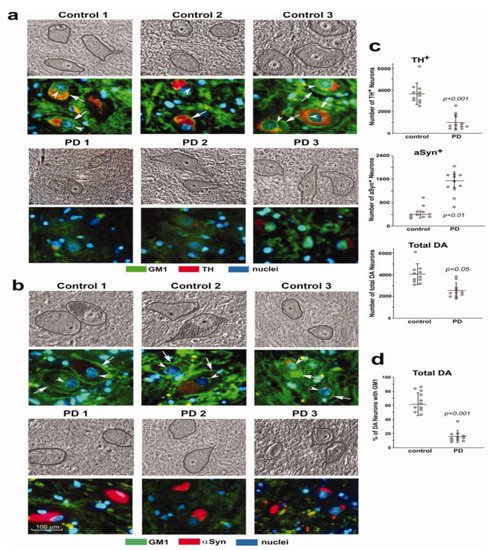
Figure 2. GM1 deficiency in SNpc neurons of PD patients. Immunohistochemical staining of GM1 and TH (a) and GM1 plus aSyn (b) in paraffin sections from SNpc region of three PD patients and three non-PD controls. Hoechst 33342-stained nuclei. DA neurons are encircled in phase images shown in upper panels. GM1 expression is indicated in both plasma membrane (arrows) and nuclear envelope (arrowheads). (c,d): Quantification for 11 sporadic PD patients and 11 non-PD controls. Eight sections of each patient were counted, with total DA neurons (TH1plus aSyn1) ranging from 3100 to 6500 in non-PD controls and from 1400 to 3800 in PD patients. Data are mean ± SD. Statistical difference was analyzed with two-tailed Student’s t-test (images used are adapted from [6] with permission granted by Journal of Neuroscience Research, Copyright 2012 Wiley Online Library).
3. Systemic Deficiency of GM1 in PD Tissues
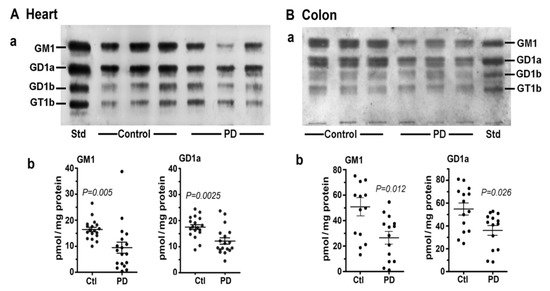
In this study [8] we first examined two tissues that are well recognized as major participants in PD pathology: colon and heart. Multiple samples of these along with age-matched controls were obtained from the Banner Sun Health Research Inst. (Sun City, AZ, USA; Dr Thomas Beach). Total lipids were extracted with chloroform (C): methanol (M) (1:1 by vol), and after volume reduction, these were applied to an HPTLC plate, which was developed in C/M/0.25M KCl, as described [7][8]. By using subunit B of cholera toxin linked to horseradish peroxidase (CtxB-HRP) to detect and quantify GM1 on the plate, we found that both the heart and colon contained significantly less GM1 than the controls (Figure 3). The same was true for GD1a, which was revealed after conversion to GM1 through the application of N’ase to the plate. Notably, it was not necessary to isolate ganglioside from the other lipids prior to thin layer analysis because most of these ran well ahead of GM1 on the plate and in any case did not react with CtxB-HRP.
Figure 3. Gangliosides in heart and colon from PD patients and age-matched controls. (A): Heart (n = 18 in each group). (B): Colon (n = 14 in each group). In (A,B), subpanel (a) is HPTLC, and subpanel (b) is densitometry quantification showing the statistical difference between PD and controls, determined by Mann–Whitney rank sum U test (images used are adapted from [8] with permission granted by Glycoconjugate J., Copyright 2021 Springer).
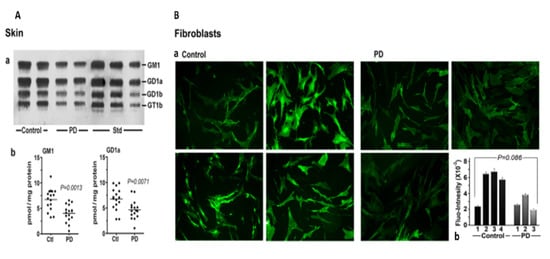
We next considered skin, a tissue somewhat less well recognized as being involved in non-motor manifestations of PD (Figure 4). Seborrheic dermatitis is one such condition, causing lesions in the head and neck along with the upper trunk and sternum. GM1 analysis via HPTLC of skin samples revealed significant deficiency for the ganglioside as well as GD1a. An analysis of cultured fibroblasts using fluorescent immunohistochemistry showed a similar trend, although this did not quite reach significance—likely due to the limited number of samples available (Figure 4B).
Figure 4. Gangliosides in skin and fibroblasts from PD patients and age-matched controls. (A): Skin (n = 16 in each group). Subpanel (a) is HPTLC, and subpanel (b) is densitometry quantification showing significant difference between PD and controls, calculated by Mann–Whitney rank sum U test. (B): (a) GM1-fluorescent images of cultured fibroblast cells from four non-PD controls and three age-matched PD patients were immunostained for GM1; (b) fluorescence intensity of GM1 staining in fibroblast cells, showing a marginal difference in GM1 by Student’s t-test (image used are adapted from [8] with permission granted by Glycoconjugate J., Copyright 2021 Springer).
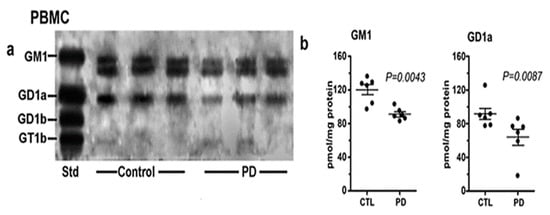
Of considerable interest was our results with PBMC (Figure 5). These white blood cells showed a similar deficiency of both GM1 and GD1a as for PD neurons, despite their lack of direct neuronal involvement. We recently carried out a more detailed study of PBMC in which we found more severe GM1 deficiency in PD patients with glucocerebrosidase malfunction than in PD patients with more ordinary sporadic PD [9]. We accordingly proposed GM1 deficiency in PBMC as a method for early diagnosis of those two forms of PD.
Figure 5. Gangliosides in PBMCs from PD patients and age-matched controls. These were analyzed with HPTLC (n = 6 in each group). Subpanel (a) is HPTLC, and subpanel (b) is densitometry quantification showing the statistical difference between PD and controls, calculated by Mann–Whitney rank sum U test (image used are adapted from [8] with permission granted by Glycoconjugate J., Copyright 2021 Springer).
The impressive array of CNS and non-CNS symptoms revealed by Braak et al. [10][11] and others more recently [12][13] is consistent with the body-wide deficiency of GM1 we are proposing. In line with that, we have suggested that GM1 deficiency constitutes a major risk factor for sporadic PD, which pertains to 90% or more of PD cases with unknown etiology. Only 10% or so of PD cases are hereditary in nature, arising from specific genetic mutations [14].
43. Cause of GM1 Deficiency
It has become recognized that a major part of the GM1 deficiency is due to the aging process itself, consistent with the discovery of Svennerholm and coworkers that both GM1 and GD1a decrease progressively with age [15][16]. However, that alone would not explain the commonly found difference between PD subjects and age-matched controls, the latter also manifesting age-related decline without developing PD. This points to the existence of one or more additional factors that further depress GM1 and GD1a in PD. One such additional influence could be defective lysosomal hydrolase, as observed in our PBMC study [9]. That study involved glucocerebrosidase, which is the most prevalent of these, but it is noteworthy that potentially damaging variants of 50 or more less prevalent lysosomal storage disorder genes have been reported in PD cases [178]. Considerable attention has been given to aSyn, the degradation of which is mainly lysosomal, and the malfunction of that process can cause aSyn accumulation and aggregation. Significantly, GM1 was shown to promote autophagy-dependent removal of aSyn in a mouse model of PD [18].
Moreover, potential influence of the microbiome has to be considered, and it was recently studied in relation to both PD model and PD subjects [19].
A fairly recent study reported significantly reduced fecal short-chain fatty acids [20], which were shown to inhibit histone deacetylase [219][2210]. Such inhibition promotes epigenetic activation of GM1 synthesis [2311][2412]. Environmental toxins are also potential inhibitors of GM1 synthesis [2513].
Finally, the recent work of Niimi et al. points to enhanced degradation of GM1 as an alternative to impaired synthesis [2614][2715]. This was based on significant downregulation of glucosylceramide in PD. The higher activity of beta-galactosidase in the blood serum of PD patients was also observed. The proposal of enhanced degradation is worth further consideration.
54. GM1 Decreases in the Periphery as well as Brain
Concerning progressive decline of GM1—due to both age and additional factors—it is important to recognize that this occurs body wide and not only in the brain. Hence, this obviates the need to postulate prion-like movement of aggregated aSyn or to debate body first vs. brain first. Such prion-like movement undoubtedly does occur but likely on a limited scale. It is difficult to observe how this would account for PD manifestations in numerous and diverse locations such as gastrointestinal, cardiovascular, and dermatological neurons among others involved in PD. Since all neurons are dependent on an adequate level of GM1 for viability and neuronal functioning, those in the periphery would be expected to suffer dysfunction—with concomitant PD symptoms—more or less in parallel with those of CNS.
An important and frequently asked question concerns the underlying biochemistry that renders GM1 so essential to neuron function and long-term viability. In general, GM1 serves several essential functions through stereospecific association with particular proteins that preserves the stereospecificity necessary for the normal functioning of those proteins. Well-established examples include nerve growth factor (NGF) and brain-derived growth factor (BDNF), both of which have receptors tightly bound to GM1 for long term preservation of neuronal functioning (discussed below). We propose that aSyn constitutes an important addition to the list of such proteins since its binding of GM1 is what holds aSyn in alpha-helical, non-aggregating conformation [2816][2917]. The latter in vitro studies have been supplemented by in vivo demonstration in PD mouse models of GM1′s ability to disperse aSyn aggregates [6][3018]. We further propose that this essential binding is carried out by the small pool of soluble GM1 [3119][3220], which was shown to bind soluble proteins (yet to be identified) [3220]. Preliminary evidence for this was obtained in the aggregation of aSyn following depletion of cytosolic GM1 in cultured NG108-15 cells (supplement to Ledeen et al., 2021) [821]. A key point is that even a small deficiency of GM1 can disrupt the homeostatic balance, resulting in increasing aSyn aggregation as the deficiency continues.
References
- Piccinini, M.; Scandroglio, F.; Prioni, S.; Buccinnà, B.; Loberto, N.; Aureli, M.; Chigorno, V.; Lupino, E.; DeMarco, G.; Lomartire, A.; et al. Deregulated Sphingolipid Metabolism and Membrane Organization in Neurodegenerative Disorders. Mol. Neurobiol. 2010, 41, 314–340.
- Ledeen, R.W.; Wu, G. Gangliosides of the Nervous System. In Methods in Molecular Biology; Sonnino, S., Prinetti, A., Eds.; Elsevier: New York, NY, USA, 2018; Volume 1804, pp. 19–25.
- Miyagi, T.; Yamaguchi, K. Mammalian sialidases: Physiological and pathological roles in cellular functions. Glycobiology 2012, 22, 880–896.
- Ledeen, R.W.; Wu, G. The multi-tasked life of GM1 ganglioside, a true factotum of nature. Trends Biochem. Sci. 2015, 40, 407–418.
- Wu, G.; Lu, Z.-H.; Kulkarni, N.; Amin, R.; Ledeen, R.W. Mice Lacking Major Brain Gangliosides Develop Parkinsonism. Neurochem. Res. 2011, 36, 1706–1714.
- Wu, G.; Lu, Z.-H.; Kulkarni, N.; Ledeen, R.W. Deficiency of ganglioside GM1 correlates with Parkinson’s disease in mice and humans. J. Neurosci. Res. 2012, 90, 1997–2008.
- Hadaczek, P.; Wu, G.; Sharma, N.; Ciesielska, A.; Bankiewicz, K.; Davidow, A.L.; Lu, Z.H.; Forsayeth, J.; Ledeen, R.W. GDNF signaling implemented by GM1 ganglioside; failure in Parkinson’s disease and GM1-deficient murine model. Exp. Neurol. 2015, 263, 177–189.
- Ledeen, R.; Chowdhury, S.; Lu, Z.H.; Chakraborty, M.; Wu, G. Systemic deficiency of GM1 ganglioside in Parkinson’s disease tissues and its relation to the disease etiology. Glycoconj. J. 2022. Robak, L.A.; Jansen, I.E.; van Rooij, J.; Uitterlinden, A.G.; Kraaij, R.; Jankovic, J.; Heutink, P.; Shulman, J.M.; Consortium, I.P.s.D.G. Excessive burden of lysosomal storage disorder gene variants in Parkinson’s disease. Brain 2017, 140, 3191–3203.
- Alselehdar, S.K.; Chakraborty, M.; Chowdhury, S.; Alcalay, R.N.; Surface, M.; Ledeen, R. Subnormal GM1 in PBMCs: Promise for Early Diagnosis of Parkinson’s Disease? Int. J. Mol. Sci. 2021, 22, 11522. Xu, W.S.; Parmigiani, R.B.; Marks, P.A. Histone deacetylase inhibitors: Molecular mechanisms of action. Oncogene 2007, 26, 5541–5552.
- Braak, H.; Del Tredici, K.; Rub, U.; De Vos, R.A.; Steur, E.N.J.; Braak, E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol. Aging. 2003, 24, 197–211. Chen, J.S.; Faller, D.V.; Spanjaard, R.A. Short-chain fatty acid inhibitors of histone deacetylases: Promising anticancer therapeutics? Curr. Cancer Drug Targets 2003, 3, 219–236.
- Braak, H.; De Vos, R.A.; Bohl, J.; Del Tredici, K. Gastric α-synuclein immunoreactive inclusions in Meissner’s and Auerbach’s plexuses in cases staged for Parkinson’s disease-related brain pathology. Neurosci. Lett. 2006, 396, 67–72. Tsai, Y.T.; Yu, R.K. Epigenetic activation of mouse ganglioside synthase genes: Implications for neurogenesis. J. Neurochem. 2014, 128, 101–110.
- Pellicano, C.; Benincasa, D.; Pisani, V.; Buttarelli, F.R.; Giovannelli, M.; Pontieri, F.E. Prodromal non-motor symptoms of Parkinson’s disease. Neuropsychiatr. Dis. Treat. 2007, 3, 145–152. Itokazu, Y.; Tsai, Y.T.; Yu, R.K. Epigenetic regulation of ganglioside expression in neural stem cells and neuronal cells. Glycoconj. J. 2017, 34, 749–756.
- Halliday, G.; Barker, R.A.; Rowe, D.B. Non-dopamine Lesions in Parkinson’s Disease; Oxford University Press: Oxford, UK, 2013. Goldman, S.M. Environmental toxins and Parkinson’s disease. Annu. Rev. Pharmacol. Toxicol. 2014, 54, 141–164.
- Bonifati, V. Genetics of parkinsonism. Parkinsonism Relat. Disord. 2007, 13 (Suppl. 3), S233–S241. Niimi, Y.; Mizutani, Y.; Akiyama, H.; Watanabe, H.; Shiroki, R.; Hirabayashi, Y.; Hoshinaga, K.; Mutoh, T. Cerebrospinal Fluid Profiles in Parkinson’s Disease: No Accumulation of Glucosylceramide, but Significant Downregulation of Active Complement C5 Fragment. J. Parkinsons Dis. 2021, 11, 221–232.
- Svennerholm, L.; Boström, K.; Jungbjer, B.; Olsson, L. Membrane lipids of adult human brain: Lipid composition of frontal and temporal lobe in subjects of age 20 to 100 years. J. Neurochem. 1994, 63, 1802–1811. Niimi, Y.; Ito, S.; Mizutani, Y.; Murate, K.; Shima, S.; Ueda, A.; Satake, W.; Hattori, N.; Toda, T.; Mutoh, T. Altered regulation of serum lysosomal acid hydrolase activities in Parkinson’s disease: A potential peripheral biomarker? Parkinsonism Relat. Disord. 2019, 61, 132–137.
- Svennerholm, L.; Boström, K.; Fredman, P.; Månsson, J.-E.; Rosengren, B.; Rynmark, B.-M. Human brain gangliosides: Developmental changes from early fetal stage to advanced age. Biochimi. Biophys. Acta 1989, 1005, 109–117. Martinez, Z.; Zhu, M.; Han, S.; Fink, A.L. GM1 specifically interacts with alpha-synuclein and inhibits fibrillation. Biochemistry 2007, 46, 1868–1877.
- Robak, L.A.; Jansen, I.E.; van Rooij, J.; Uitterlinden, A.G.; Kraaij, R.; Jankovic, J.; Heutink, P.; Shulman, J.M.; Consortium, I.P.s.D.G. Excessive burden of lysosomal storage disorder gene variants in Parkinson’s disease. Brain 2017, 140, 3191–3203. Bartels, T.; Kim, N.C.; Luth, E.S.; Selkoe, D.J. N-alpha-acetylation of α-synuclein increases its helical folding propensity, GM1 binding specificity and resistance to aggregation. PLoS ONE 2014, 9, e103727.
- Guo, Y.L.; Duan, W.J.; Lu, D.H.; Ma, X.H.; Li, X.X.; Li, Z.; Bi, W.; Kurihara, H.; Liu, H.Z.; Li, Y.F.; et al. Autophagy-dependent removal of α-synuclein: A novel mechanism of GM1 ganglioside neuroprotection against Parkinson’s disease. Acta Pharmacol. Sin. 2021, 42, 518–528. Schneider, J.S.; Aras, R.; Williams, C.K.; Koprich, J.B.; Brotchie, J.M.; Singh, V. GM1 Ganglioside Modifies α-Synuclein Toxicity and is Neuroprotective in a Rat α-Synuclein Model of Parkinson’s Disease. Sci. Rep. 2019, 9, 8362.
- Keshavarzian, A.; Engen, P.; Bonvegna, S.; Cilia, R. Chapter 11—The gut microbiome in Parkinson’s disease: A culprit or a bystander? In Progress in Brain Research; Björklund, A., Cenci, M.A., Eds.; Elsevier: Amsterdam, The Netherlands, 2020; Volume 252, pp. 357–450. Ledeen, R.W.; Skrivanek, J.A.; Tirri, L.J.; Margolis, R.K.; Margolis, R.U. Gangliosides of the neuron: Localization and origin. Adv Exp. Med. Biol. 1976, 71, 83–103.
- Unger, M.M.; Spiegel, J.; Dillmann, K.U.; Grundmann, D.; Philippeit, H.; Bürmann, J.; Faßbender, K.; Schwiertz, A.; Schäfer, K.H. Short chain fatty acids and gut microbiota differ between patients with Parkinson’s disease and age-matched controls. Parkinsonism Relat. Disord. 2016, 32, 66–72. Sonnino, S.; Ghidoni, R.; Fiorilli, A.; Venerando, B.; Tettamanti, G. Cytosolic gangliosides of rat brain: Their fractionation into protein-bound complexes of different ganglioside compositions. J. Neurosci. Res. 1984, 12, 193–204.
- Xu, W.S.; Parmigiani, R.B.; Marks, P.A. Histone deacetylase inhibitors: Molecular mechanisms of action. Oncogene 2007, 26, 5541–5552. Ledeen, R.; Chowdhury, S.; Lu, Z.H.; Chakraborty, M.; Wu, G. Systemic deficiency of GM1 ganglioside in Parkinson’s disease tissues and its relation to the disease etiology. Glycoconj. J. 2022.
- Chen, J.S.; Faller, D.V.; Spanjaard, R.A. Short-chain fatty acid inhibitors of histone deacetylases: Promising anticancer therapeutics? Curr. Cancer Drug Targets 2003, 3, 219–236.
- Tsai, Y.T.; Yu, R.K. Epigenetic activation of mouse ganglioside synthase genes: Implications for neurogenesis. J. Neurochem. 2014, 128, 101–110.
- Itokazu, Y.; Tsai, Y.T.; Yu, R.K. Epigenetic regulation of ganglioside expression in neural stem cells and neuronal cells. Glycoconj. J. 2017, 34, 749–756.
- Goldman, S.M. Environmental toxins and Parkinson’s disease. Annu. Rev. Pharmacol. Toxicol. 2014, 54, 141–164.
- Niimi, Y.; Mizutani, Y.; Akiyama, H.; Watanabe, H.; Shiroki, R.; Hirabayashi, Y.; Hoshinaga, K.; Mutoh, T. Cerebrospinal Fluid Profiles in Parkinson’s Disease: No Accumulation of Glucosylceramide, but Significant Downregulation of Active Complement C5 Fragment. J. Parkinsons Dis. 2021, 11, 221–232.
- Niimi, Y.; Ito, S.; Mizutani, Y.; Murate, K.; Shima, S.; Ueda, A.; Satake, W.; Hattori, N.; Toda, T.; Mutoh, T. Altered regulation of serum lysosomal acid hydrolase activities in Parkinson’s disease: A potential peripheral biomarker? Parkinsonism Relat. Disord. 2019, 61, 132–137.
- Martinez, Z.; Zhu, M.; Han, S.; Fink, A.L. GM1 specifically interacts with alpha-synuclein and inhibits fibrillation. Biochemistry 2007, 46, 1868–1877.
- Bartels, T.; Kim, N.C.; Luth, E.S.; Selkoe, D.J. N-alpha-acetylation of α-synuclein increases its helical folding propensity, GM1 binding specificity and resistance to aggregation. PLoS ONE 2014, 9, e103727.
- Schneider, J.S.; Aras, R.; Williams, C.K.; Koprich, J.B.; Brotchie, J.M.; Singh, V. GM1 Ganglioside Modifies α-Synuclein Toxicity and is Neuroprotective in a Rat α-Synuclein Model of Parkinson’s Disease. Sci. Rep. 2019, 9, 8362.
- Ledeen, R.W.; Skrivanek, J.A.; Tirri, L.J.; Margolis, R.K.; Margolis, R.U. Gangliosides of the neuron: Localization and origin. Adv Exp. Med. Biol. 1976, 71, 83–103.
- Sonnino, S.; Ghidoni, R.; Fiorilli, A.; Venerando, B.; Tettamanti, G. Cytosolic gangliosides of rat brain: Their fractionation into protein-bound complexes of different ganglioside compositions. J. Neurosci. Res. 1984, 12, 193–204.
- Mutoh, T.; Tokuda, A.; Miyadai, T.; Hamaguchi, M.; Fujiki, N. Ganglioside GM1 binds to the Trk protein and regulates receptor function. Proc. Natl. Acad. Sci. USA 1995, 92, 5087–5091.
- Pitto, J.; Mutoh, T.; Kuriyama, M.; Ferraretto, A.; Palestini, P.; Masserini, M. Influence of endogenous GM1 ganglioside on TrkB activity in cultured neurons. FEBS Lett. 1998, 439, 93–96.
- Pascual, A.; Hidalgo-Figueroa, M.; Piruat, J.I.; Pintado, C.O.; Gómez-Díaz, R.; López-Barneo, J. Absolute requirement of GDNF for adult catecholaminergic neuron survival. Nat. Neurosci. 2008, 11, 755–761.
- Chiricozzi, E.; Lunghi, G.; Di Biase, E.; Fazzari, M.; Sonnino, S.; Mauri, L. GM1 Ganglioside Is A Key Factor in Maintaining the Mammalian Neuronal Functions Avoiding Neurodegeneration. Int. J. Mol. Sci. 2020, 21, 868.
- Ledeen, R.W.; Wu, G. Gangliosides, α-Synuclein, and Parkinson’s Disease. In Progress in Molecular Biology and Translational Science; Schnaar, R.L., Lopez, P.H.H., Eds.; Academic Press: Cambridge, MA, USA, 2018; Volume 156, pp. 435–454.
- Schneider, J.S.; Sendek, S.; Daskalakis, C.; Cambi, F. GM1 ganglioside in Parkinson’s disease: Results of a five year open study. J. Neurol. Sci. 2010, 292, 45–51.
- Schneider, J.S.; Gollomp, S.M.; Sendek, S.; Colcher, A.; Cambi, F.; Du, W. A randomized, controlled, delayed start trial of GM1 ganglioside in treated Parkinson’s disease patients. J. Neurol. Sci. 2013, 324, 140–148.
- Wu, G.; Lu, Z.H.; Seo, J.H.; Alselehdar, S.K.; DeFrees, S.; Ledeen, R.W. Mice deficient in GM1 manifest both motor and non-motor symptoms of Parkinson’s disease; successful treatment with synthetic GM1 ganglioside. Exp. Neurol. 2020, 329, 113284.
More