Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 3 by Dean Liu and Version 4 by Dean Liu.
Avian orthoavulaviruses type-1 (AOaV-1) have transitioned from animal vaccine vector to a bona fide vaccine delivery vehicle in human. Owing to induction of robust innate and adaptive immune responses in mucus membranes in both birds and mammals, AOaVs offer an attractive vaccine against respiratory pathogens. The unique features of AOaVs include over 50 years of safety profile, stable expression of foreign genes, high infectivity rates in avian and mammalian hosts, broad host spectrum, limited possibility of recombination and lack of pre-existing immunity in humans. Additionally, AOaVs vectors allow the production of economical and high quantities of vaccine antigen in chicken embryonated eggs and several GMP-grade mammalian cell lines.
- avian orthoavulaviruses type-1
- human
- animal
- vaccines
1. Introduction
Avian orthoavulavirus type-1 (AOaV-1), previously known as Newcastle disease virus (NDV), is a non-segmented negative-sense RNA virus belonging to paramyxovirus, which naturally infects birds, causing severe economic losses globally [1]. Although AOaV-1 is naturally monotypic, antigenic and genetic diversity have been reported among AOaV-1 isolates. Two distinct classification methods for AOaV-1 are now in use worldwide. When comparing the isolated sequences, one of the systems separates AOaV-1 into two primary divisions, Class I and Class II. Class I is further subdivided into nine genotypes while class II is subdivided into ten genotypes. Class II viruses have been studied in detail, and the early 1930–1960 genotypes I, II, III, IV, and IX have 15,186 nucleotides. [2][3][4]. Genotypes V, VI, VII, VIII, and X that appeared late (after 1960) comprise 15,192 nucleotides.
Live vaccines based on lentogenic AOaV-1 strains Hitchner B1, LaSota, Fuller (F), and mesogenic strain R2B are commonly utilised. AOaV-1 strain F is a low-virulence virus that has been investigated in different countries throughout Europe, Africa, and Asia in the form of a live vaccine [5]. AOaV-1 research is not only focused on its pathobiology, but also on its potential use as a vaccine vector to develop novel vaccines for poultry and mammals, including humans [6][7][8]. Numerous AOaV-1-vectored vaccines expressing protective antigens from various pathogens have been developed since the first recombinant AOaV-1 expressing a foreign gene in 2000 (Figure 1) [9]. In addition, AOaV-1 encodes six major proteins, resulting in less immune response competition between vector proteins and the expressed foreign antigen. The AOaV-1 replicates in the cytoplasm do not integrate into the host cellular DNA, and do not cause persistent infection. In addition, the AOaV-1-vectored vaccine can also be used as a “differentiating infected from vaccinated animals” (DIVA) vaccine. Therefore, the aim is to summarise the recent advances in the field of the AOaV-1 vectored vaccine and discuss its potential application against emerging and re-emerging animal and human viral pathogens.
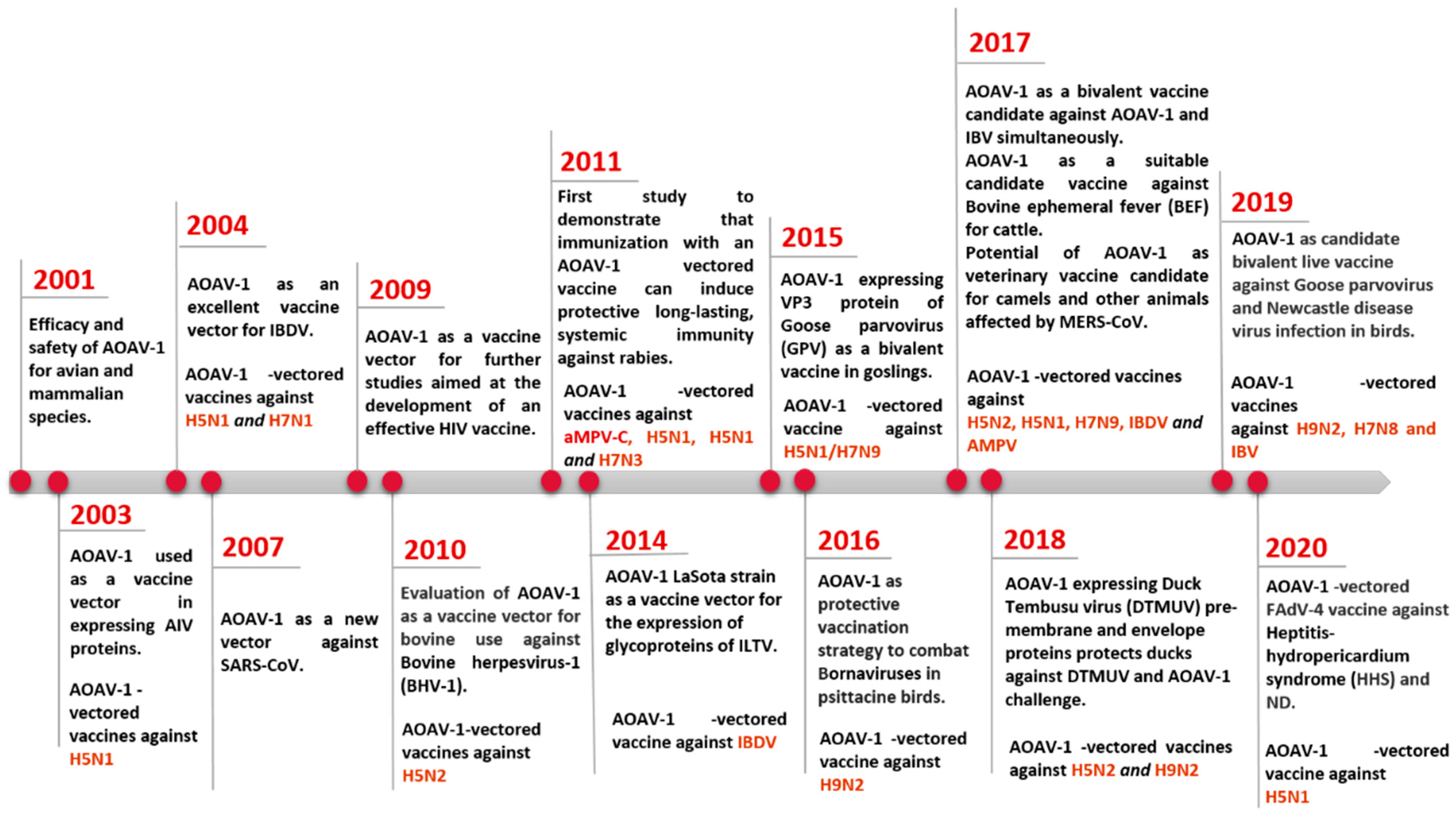
Figure 1. Progress on the use of AOaV-1 as viral vector for vaccine development.
2. Genomic and Biological Features of Avian Orthoavulavirus Type-1
The AOaV-1 causes one of the most significant avian viral diseases, responsible for severe economic losses globally [10]. AOaV-1 is taxonomically classified a member of the order Mononegavirales, family Paramyxoviridae, sub-family Paramoxivirinae under genus Avulavirus [10][11]. The AOaV-1 virion structure is pleomorphic but mostly roughly spherical with diameters of around 100–500 nm [11][12]. The AOaV-1 genome is around 15 kb of linear single-stranded, negative-sense RNA with six essential genes from 3′ to 5′ direction- nucleocapsid (N), phosphoprotein (P), matrix protein (M), fusion protein (F), haemagglutinin-neuraminidase protein (HN), and large polymerase protein (L) [13]. Each gene is identified by the presence of signal sequences at the start (GS—gene-start) and at the end of the gene (GE—gene-end). Additionally, the presence of intergenic sequences (IGS) separates the genes from each other [11]. All AOaV-1 genes code for a single major protein; however, due to transcriptional editing or differential initiation of gene P, mRNA may result in the formation of a non-structural V and/or W protein [13]. The viral RNA polymerase (L protein) initiates the transcription of the viral genome at the 3′ terminal nucleotide, sequentially terminating and reinitiating at multiple start/stop gene sites. At the end of each gene, a fraction of RNA polymerase dissociates from the genome and is unable to restart transcription to the next downstream gene, thus resulting in diminished quantities of mRNAs for genes located further the 3′ end of the genome. This mechanism enables the balance in synthesising proteins that are needed in high concentration such as N and M and proteins needed in smaller amounts (i.e., HN and L) (Figure 2) [13].
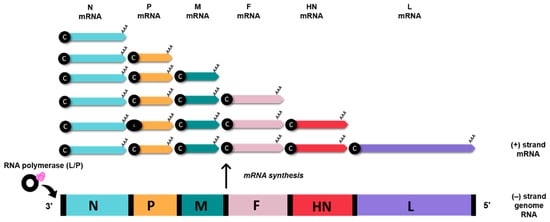
Figure 2. AOaV-1 mRNA synthesis. Viral (-) strand genome serves as templates for the generation of sub-genomic mRNAs. The RNA polymerase (L/P) initiates mRNA synthesis at the beginning of the N gene, near the 3′ end of the viral genome. Small black spheres denote 5’ cap (c), AAA denotes poly (A) tail.
Virulence and infectivity of AOaV-1 depends on both F and HN proteins [14][15]. Co-expression and interaction of homologous F and HN proteins are required for virus fusion. The HN protein is involved in virus-specific membrane fusion. Through haemagglutination and neuraminidase activities, the HN protein facilitates the interaction between host receptors and virus, which allows the host membrane and viral membrane to be closer together, granting the F protein to come in contact with the host cell, so the virus will be able to penetrate the cell surface [12]. The F protein mediates the fusion of the AOaV-1 with the host cell that undergoes a conformational change, allowing the cleavage of the protein precursor F0 to F1/F2 subunits, which also brings the viral envelope closer to the cell membrane, leading to the fusion of the two membranes [13]. Thus, the amino acid sequence of the cleavage site of the F protein is theorised to be a major determinant of infection [11][14][16].
3. AOaV-1 as a Viral Vaccine Vector
Recent advancements in recombinant DNA technologies, molecular biology, and biotechnology provided the platform to further understand AOaV-1 as a viral pathogen, as well as its potential to serve as vaccine and or vaccine vector for humans and animals. Due to the modular nature of transcription, the low recombination frequency, and the absence of DNA phase during replication, AOaV-1 is considered a promising candidate for the development of live attenuated vaccines and vaccine vectors [11]. Using reverse genetics, AOaV-1 can be easily engineered to bear the desired antigens as additional proteins, by using a recombinant viruses recovered from cloned cDNA (Figure 3) [17][18][19][20]. The benefits of AOaV-1 as a viral vector include (1) its ability to replicate efficiently in vivo with high titers; (2) its ability to elicit humoral and cellular immune cell responses; (3) its ease of genome manipulation; (4) its independent replication and failure to integrate into the host genome; and (5) stable expression of foreign proteins [11][16][19].
Figure 3. Diagrammatic representation of the reverse genetics strategy for AOAV-1 rescue. Construction of recombinant AOAV-1 containing foreign gene (A). The recombinant AOAV-1 will be co-transfected together with expression helper plasmids encoding the N, P, and L proteins of AOAV-1 and infected with recombinant vaccinia MVA/T7 expressing T7 RNA polymerase (B).
The transfection of cultured cells with plasmids encoding the viral components of functional nucleocapsid, full-length antigenomic RNA, major proteins involved in viral replication and transcription (N, P and L) under the control of bacteriophage T7 RNA polymerase promoter, allows infectious AOaV-1 to be recovered entirely from cloned cDNA, which is also known as reverse genetic technique (Figure 3) [16][17][21][22][23][24]. A foreign gene flanked by AOaV-1 GS and GE sequences was inserted within a non-coding region of the AOaV-1 genome. Foreign genes are more efficiently expressed when they are inserted closer to the 3′ end of the genome due to polar gradient transcription. While it is possible to insert a foreign gene between any two AOaV-1 genes, i.e., the insertion site between the P and M genes has proved ideal for efficient expression of the extraneous proteins [24][6][1][2]. The AOaV-1 adapts the external genes with good stability and can express up to three foreign genes within a single AOaV-1 vector [16].
LaSota and B1 lentogenic AOaV-1 strains were commonly used as vaccine vectors due to their safety records. Mesogenic and velogenic AOaV-1 strains are not being used in chickens as vaccine vector due to their virulence in chickens. An in vivo study for a AOaV-1 vectored vaccine in non-human primates based on the mesogenic strain (Beaudette C) revealed its efficient replication with a substantially higher level of antibodies compared to the LaSota strain vectored vaccine [3], indicating that it would be an efficient vaccine vector.
4. Application of AOaV-1 as Vaccine Vector for Poultry and Animal Viruses
Efforts for studying the AOaV-1 as a viral vector have given rise to various vaccine candidates expressing antigens of various viral pathogens in poultry, animals, and humans [4]. Preclinical and clinical studies have been conducted to assess the safety, immunogenicity, and protective efficacy of these AOaV-1-vectored vaccines.5. Current Application of AOaV-1 as Vaccine Vector for Emerging and Remerging Human Viruses
Conventional vaccines have effectively reduced the burden for many infectious disease, i.e., small pox eradication and substantially controlling diseases such as polio, tetanus, diphtheria, and measles [25]. Live attenuated viruses, inactivated viruses, or recombinant subunit-based vaccines are traditional platforms for the development of vaccines, which often contribute to a long-term immunity of many infectious human and/or animal viruses. However, a majority of them are not suitable for human use due to safety concerns, poor efficacy, or easy impracticability, and are not always suitable or even feasible in outbreak situations [25]. Additionally, scenarios of outbreaks may limit the development and or productivity of conventional vaccines. A number of challenges need to be overcome to prove the efficacy of these conventional vaccines in the face of an emerging or future pandemic [25]. One of the key problems for pandemic preparedness is the unpredictable nature of emerging pathogens and zoonosis that poses a permanent threat to the population, as with SARS-CoV-2. SARS-CoV-2 outbreaks revealed the possibility of the known pathogens for mutations and adaptation to a new host or environment with impermissible consequences for their immunogenic properties and the seriousness of the symptoms they produce. The risk of such events is high in RNA viruses, whose high mutation rates favour adaptability, as demonstrated by recent epidemics and pandemics. Since the goals of the vaccine remain undefined before an outbreak, time remains an important obstacle to effective development of the vaccine. The average development time currently exceeds 10 years for conventional preclinical vaccines [26], highlighting the dire need for new approaches which permit extremely rapid development and licencing to prevent the emerging outbreak from spreading worldwide. Another major problem is the cost of vaccine development and production; the development of a new vaccine candidate with established technologies is estimated to exceed USD 500 million with additional costs for setting up equipment and facilities between USD 50 million and 700 million [27]. Although some vaccine development costs cannot be avoided to meet the necessary safety standards, validation and production costs are high in every vaccine requirement for dedicated manufacturing processes and facilities in most conventional vaccine technologies. In addition, new technologies are required in order to support more cost-efficient vaccination production, particularly in light of resource limited environments and the fact that emergencies represent niche markets. The second problem is the manufacturing capacity of established methods, often inadequate for global vaccination. Although the potential threat is recognised, vaccine production technology, such as the COVID-19 vaccine, is still problematic in its production capacity to meet peak demands of a disease. An example is that the potential influenza pandemic vaccine production capacity could theatrically support vaccination of 43% of the population with two doses of the vaccine in 2015 through WHO efforts [28]. In 2015, only 5% of influenza vaccine doses were distributed to South-east Asia, the Eastern Mediterranean, and Africa in the WHO regions, which represent about half of the world’s population, but the distribution of vaccine products worldwide is far from equal between the developing and industrialised countries [29]. Moreover, most currently authorised vaccines would require 3–5 months from virus identification to the distribution of vaccines, providing the virus with ample time to spread globally. Therefore, in the event of pandemic risks, technology that allows quick manufacturing of a large number of vaccines is absolutely necessary. The use of recombinant vectors as a vaccination tool for human pathogens was therefore critical, due to their ability to express high-level foreign proteins in host cells, which leads to a strong, long-term immune response to the target protein. As AOaV-1 is an avian paramyxovirus, a major advantage of the vaccine platform is that the issue of anti-vector pre-existing immunity is not considered a major factor. Further, several AOaV-1 strains are licensed and readily available for use as veterinary vaccines. Although even small exogenous transgenes may significantly lower the yield of recombinant viruses, most recombinant AOaV-1 vectors can be propagated to high titers in chicken eggs and even some cell lines [17]. Recombinant AOaV-1 has also been used as a viral vector in the delivery of vaccine antigen for humans [30]. Currently, AOaV-1 vaccine candidates both respiratory and non-respiratory diseases include SARS-CoV-2 [24][31], SARS-CoV [32], EBOV [33], HIV-1 [6][34], HPAIV H5N1 [8], RSV [35], and HPIV-3 [3] (Table 1).Table 1. Candidate AOaV-1-vectored vaccines for human use.
| Pathogen | AOaV-1 Backbone | Antigen | Insert Site | Animal Model | Vaccination (Route) | References |
|---|---|---|---|---|---|---|
| HIV-1 | Hitchner B1 | Gag | P/M | Mouse | i.n. | [6] |
| HIV-1 | La Sota | Gag | P/M | Mouse | i.n. | [36] |
| HIV-1 | La Sota | Gag; Env; Gag + Env | Env- P/M and Gag- HN/L; Gag- O/M and Env- HN/L; Env + Gag- P/M; Env- P/M; Gag- P/M | Guinea pigs/Mouse | i.n. | [34] |
| SIV | La Sota | gp160 | P/M | Guinea pigs/Mouse | i.n. | [37] |
| EBOV | Beaudette C and La Sota | GP | P/M | Rhesus monkeys | i.n/i.t. | [33] |
| EBOV | Chimeric AOaV-1 | GP | N/P, P/M, and M/F | Guinea pigs | i.n. | [38] |
| HPIV-3 | Beaudette C and La Sota | HN | P/M | African green monkeys and rhesus monkeys | i.n./i.t. | [3] |
| RSV | Hitchner B1 | F | P/M | Mouse | i.n. | [35] |
| Poliovirus | La Sota | P1 and 3CD | P1- P/M and 3CD- HN/L | Guinea pigs | i.n. | [39] |
| Lyme | LaSota/VF | BmpA + OspC | P/M | Hamsters | i.n./i.m./i.p. | [40] |
Human immunodeficiency virus-1 [HIV-1]; simian immunodeficiency virus (SIV); Ebola virus (EBOV); human parainfluenza virus type-3 (HPIV-3); respiratory syncytial virus (RSV); group specific antigen (Gag); envelope glycoprotein gp160 (Env); envelope protein gp160 (gp160); (GP); hemagglutinin (HN); fusion (F); capsid protein precursor (P1); viral protease (3CD); basic membrane protein A (BmpA); outer surface protein C (OspC); phosphoprotein (P); matrix (M); large polymerase (L); nucleocapsid (N); intranasal (i.n.); intratumoral (i.t.); intraperitoneal (i.p.); intramuscular (i.m.).
References
- Nakaya, T.; Cros, J.; Park, M.-S.; Nakaya, Y.; Zheng, H.; Sagrera, A.; Villar, E.; García-Sastre, A.; Palese, P. Recombinant Newcastle disease virus as a vaccine vector. J. Virol. 2001, 75, 11868–11873.
- Zhao, H.; Peeters, B.P.H. Recombinant Newcastle disease virus as a viral vector: Effect of genomic location of foreign gene on gene expression and virus replication. J. Gen. Virol. 2003, 84, 781–788.
- Bukreyev, A.; Huang, Z.; Yang, L.; Elankumaran, S.; St. Claire, M.; Murphy, B.R.; Samal, S.K.; Collins, P.L. Recombinant Newcastle disease virus expressing a foreign viral antigen is attenuated and highly immunogenic in primates. J. Virol. 2005, 79, 13275–13284.
- Hu, Z.; Ni, J.; Cao, Y.; Liu, X. Newcastle disease virus as a vaccine vector for 20 years: A focus on maternally derived antibody interference. Vaccines 2020, 8, 222.
- Choi, K.S. Newcastle disease virus vectored vaccines as bivalent or antigen delivery vaccines. Clin. Exp. Vaccine Res. 2017, 6, 72–82.
- Carnero, E.; Li, W.; Borderia, A.V.; Moltedo, B.; Moran, T.; García-Sastre, A. Optimization of human immunodeficiency virus gag expression by Newcastle disease virus vectors for the induction of potent immune responses. J. Virol. 2009, 83, 584–597.
- Tan, L.; Wen, G.; Qiu, X.; Yuan, Y.; Meng, C.; Sun, Y.; Liao, Y.; Song, C.; Liu, W.; Shi, Y.; et al. A recombinant la sota vaccine strain expressing multiple epitopes of infectious bronchitis virus (Ibv) protects specific pathogen-free (spf) chickens against ibv and ndv challenges. Vaccines 2019, 7, 170.
- DiNapoli, J.M.; Yang, L.; Suguitan, A.; Elankumaran, S.; Dorward, D.W.; Murphy, B.R.; Samal, S.K.; Collins, P.L.; Bukreyev, A. Immunization of primates with a Newcastle disease virus-vectored vaccine via the respiratory tract induces a high titer of serum neutralizing antibodies against highly pathogenic avian influenza virus. J. Virol. 2007, 81, 11560–11568.
- Kim, S.H.; Samal, S.K. Innovation in newcastle disease virus vectored avian influenza vaccines. Viruses 2019, 11, 300.
- Alexander, D.J. Newcastle disease. Br. Poultry Sci. 2001, 42, 5–22.
- Ganar, K.; Das, M.; Sinha, S.; Kumar, S. Newcastle disease virus: Current status and our understanding. Virus Res. 2014, 184, 71–81.
- Yusoff, K.; Tan, W.S. Newcastle disease virus: Macromolecules and opportunities. Avian Pathol. 2001, 30, 439–455.
- Acheson, N.H.; Kolakofsky, D.; Richardson, C. Paramyxoviruses and Rhabdoviruses. In Fundamentals of Molecular Virology; Acheson, N.H., Ed.; Wiley: Hoboken, NJ, USA, 2011; pp. 175–187.
- Panda, A.; Huang, Z.; Elankumaran, S.; Rockemann, D.D.; Samal, S.K. Role of fusion protein cleavage site in the virulence of Newcastle disease virus. Microb. Pathog. 2004, 36, 1–10.
- Iorio, R.M.; Mahon, P.J. Paramyxoviruses: Different receptors—Different mechanisms of fusion. Trends Microbiol. 2008, 16, 135–137.
- Samal, S.; Kumar, S.; Khattar, S.K.; Samal, S.K. A single amino acid change, Q114R, in the cleavage-site sequence of Newcastle disease virus fusion protein attenuates viral replication and pathogenicity. J. Gen. Virol. 2011, 92, 2333–2338.
- Krishnamurthy, S.; Huang, Z.; Samal, S.K. Recovery of a virulent strain of Newcastle disease virus from cloned cDNA: Expression of a foreign gene results in growth retardation and attenuation. Virology 2000, 278, 168–182.
- Bashir Bello, M.; Yusoff, K.; Ideris, A.; Hair-Bejo, M.; Hassan Jibril, A.; Peeters, B.P.H.; Omar, A.R. Exploring the prospects of engineered Newcastle disease virus in modern vaccinology. Viruses 2020, 12, 1–23.
- Huang, Z.; Elankumaran, S.; Panda, A.; Samal, S.K. Recombinant Newcastle disease virus as a vaccine vector. Poultry Sci. 2003, 82, 899–906.
- Peeters, B.P.H.; De Leeuw, O.S.; Verstegen, I.; Koch, G.; Gielkens, A.L.J. Generation of a recombinant chimeric Newcastle disease virus vaccine that allows serological differentiation between vaccinated and infected animals. Vaccine 2001, 19, 1616–1627.
- Huang, Z.; Krishnamurthy, S.; Panda, A.; Samal, S.K. High-level expression of a foreign gene from the most 3’-proximal locus of a recombinant Newcastle disease virus. J. Gen. Virol. 2001, 82, 1729–1736.
- Xiao, S.; Nayak, B.; Samuel, A.; Paldurai, A.; Kanabagattebasavarajappa, M.; Prajitno, T.Y.; Bharoto, E.E.; Collins, P.L.; Samal, S.K. Generation by reverse genetics of an effective, stable, live-attenuated Newcastle disease virus vaccine based on a currently circulating, highly virulent indonesian strain. PLoS ONE 2012, 7, e52751.
- Paldurai, A.; Kim, S.-H.; Nayak, B.; Xiao, S.; Shive, H.; Collins, P.L.; Samal, S.K. Evaluation of the contributions of individual viral genes to Newcastle disease virus virulence and pathogenesis. J. Virol. 2014, 88, 8579–8596.
- Rohaim, M.A.; Munir, M. A Scalable Topical Vectored Vaccine Candidate against SARS-CoV-2. Vaccines 2020, 8, 472.
- Rauch, S.; Jasny, E.; Schmidt, K.E.; Petsch, B. New vaccine technologies to combat outbreak situations. Front. Immunol. 2018, 9, 1963.
- Pronker, E.S.; Weenen, T.C.; Commandeur, H.; Claassen, E.H.J.H.M.; Osterhaus, A.D.M.E. Risk in vaccine research and development quantified. PLoS ONE 2013, 8, e57755.
- Plotkin, S.; Robinson, J.M.; Cunningham, G.; Iqbal, R.; Larsen, S. The complexity and cost of vaccine manufacturing—An overview. Vaccine 2017, 35, 4064–4071.
- McLean, K.A.; Goldin, S.; Nannei, C.; Sparrow, E.; Torelli, G. The 2015 global production capacity of seasonal and pandemic influenza vaccine. Vaccine 2016, 34, 5410–5413.
- Gilbert, J.A. Seasonal and pandemic influenza: Global fatigue versus global preparedness. Lancet Respir. Med. 2018, 6, 94–95.
- Kim, S.H.; Samal, S.K. Newcastle disease virus as a vaccine vector for development of human and veterinary vaccines. Viruses 2016, 8, 183.
- Sun, W.; Leist, S.R.; McCroskery, S.; Liu, Y.; Slamanig, S.; Oliva, J.; Amanat, F.; Schäfer, A.; Dinnon, K.H., 3rd; García-Sastre, A.; et al. Newcastle disease virus (NDV) expressing the spike protein of SARS-CoV-2 as a live virus vaccine candidate. EBioMedicine 2020, 62, 103132.
- DiNapoli, J.M.; Kotelkin, A.; Yang, L.; Elankumaran, S.; Murphy, B.R.; Samal, S.K.; Collins, P.L.; Bukreyev, A. Newcastle disease virus, a host range-restricted virus, as a vaccine vector for intranasal immunization against emerging pathogens. Proc. Natl. Acad. Sci. USA 2007, 104, 9788–9793.
- DiNapoli, J.M.; Yang, L.; Samal, S.K.; Murphy, B.R.; Collins, P.L.; Bukreyev, A. Respiratory tract immunization of non-human primates with a Newcastle disease virus-vectored vaccine candidate against Ebola virus elicits a neutralizing antibody response. Vaccine 2010, 29, 17–25.
- Khattar, S.K.; Manoharan, V.; Bhattarai, B.; Labranche, C.C.; Montefiori, D.C.; Samal, S.K. Mucosal immunization with newcastle disease virus vector coexpressing HIV-1 env and gag proteins elicits potent serum, mucosal, and cellular immune responses that protect against vaccinia virus env and gag challenges. MBio 2015, 6, 1–10.
- Martinez-Sobrido, L.; Gitiban, N.; Fernandez-Sesma, A.; Cros, J.; Mertz, S.E.; Jewell, N.A.; Hammond, S.; Flano, E.; Durbin, R.K.; García-Sastre, A.; et al. Protection against respiratory syncytial virus by a recombinant Newcastle disease virus Vector. J. Virol. 2006, 80, 1130–1139.
- Maamary, J.; Array, F.; Gao, Q.; Garcia-Sastre, A.; Steinman, R.M.; Palese, P.; Nchinda, G. Newcastle disease virus expressing a dendritic cell-targeted HIV gag protein induces a potent gag-specific immune response in mice. J. Virol. 2011, 85, 2235–2246.
- Manoharan, V.K.; Khattar, S.K.; Labranche, C.C.; Montefiori, D.C.; Samal, S.K. Modified Newcastle Disease virus as an improved vaccine vector against Simian Immunodeficiency virus. Sci. Rep. 2018, 8, 8952.
- Yoshida, A.; Kim, S.H.; Manoharan, V.K.; Varghese, B.P.; Paldurai, A.; Samal, S.K. Novel avian paramyxovirus-based vaccine vectors expressing the Ebola virus glycoprotein elicit mucosal and humoral immune responses in guinea pigs. Sci. Rep. 2019, 9, 5520.
- Viktorova, E.G.; Khattar, S.K.; Kouiavskaia, D.; Laassri, M.; Zagorodnyaya, T.; Dragunsky, E.; Samal, S.; Chumakov, K.; Belov, G.A. Newcastle disease virus-based vectored vaccine against Poliomyelitis. J. Virol. 2018, 92, e00976-18.
- Xiao, S.; Kumar, M.; Yang, X.; Akkoyunlu, M.; Collins, P.L.; Samal, S.K.; Pal, U. A host-restricted viral vector for antigen-specific immunization against Lyme disease pathogen. Vaccine 2011, 29, 5294–5303.
More