Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Davide Leardini and Version 2 by Rita Xu.
CBL mutations are progressively being described as involved in different clinical manifestations. Somatic CBL mutations can be found in different type of cancer. The clinical spectrum of germline mutations configures the so-called CBL syndrome, a cancer-predisposing condition that includes multisystemic involvement characterized by variable phenotypic expression and expressivity.
- CBL
- CBL syndrome
- cancer predisposition
1. Introduction
Over the last years, large-scale genomic studies have uncovered a genetic predisposition in a large variety of cancers, especially in children [1]. A recent study based on The Cancer Genome Atlas (TCGA) data set reported that approximately 8% of adult patients with cancer have a germline predisposing mutation [2]. A similar prevalence of germline mutations in cancer-predisposing genes has been identified in children and adolescents with cancer [3]. In addition to providing important insights into diagnostic and prognostic information, the evaluation of non-tumor or germline material has provided novel information on previously unrecognized non-malignant clinical spectrum [4][5][6][7][8][4,5,6,7,8]. Casitas B lineage lymphoma (CBL) is a multifunctional gene codifying for a class of adaptor proteins (CBL family proteins) implicated in the regulation of signal transduction in many physiological and pathological processes. Particularly, CBL has been reported to play a role in different well-known cell pathways, many of those are related to cancer onset and progression, hematopoietic development and T cell receptor regulation. Growing evidence have defined the clinical spectrum of germline CBL mutations configuring the so-called CBL syndrome, a cancer-predisposing condition that has been shown to also include multisystemic diseases.
2. Structure and Function of CBL
CBL proteins are included in a highly conserved family of RING finger (RF) ubiquitin ligases (E3s). Three CBL member proteins have been described: CBL, CBL-b and CBL-c [9]. CBL and CBL-b are structurally more related, while CBL-c differs by lacking the C-terminal domains. The structure of CBL protein comprises an N-terminal tyrosine kinase binding (TKB) domain followed by a linker region, a RING domain, a proline-rich region (PRR), a C-terminal phosphorylation site and a ubiquitin-association domain (Figure 1).
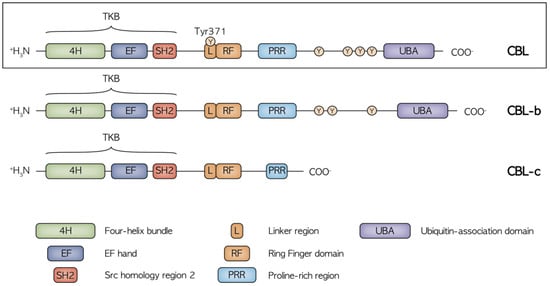
Figure 1. Structure of the CBL, CBL-b and CBL-c proteins with the relative functional domains and main phosphorylation sites. CBL and CBL-b are structurally related while CBL-c lacks the C terminal domain, including the UBA region and the proline and tyrosine rich region, mediating the binding to the SH2 and SH3 domain-containing proteins. The highly conserved N terminal region with the TKB, Linker and RF domains is crucial in the regulation of receptor and non-receptor tyrosine kinase activity.
The main known function of CBL proteins is the regulation of receptor and non-receptor tyrosine kinases (RTKs and non-RTKs, respectively), which is implicated in the transduction of signals from immune receptors (T-cell receptor, B-cell receptors and Fc receptors). It has been demonstrated that CBL proteins negatively regulate RTKs and non-RTKs proteins through their ubiquitination and degradation [9][10][9,10], mediating the transfer of ubiquitin from E2 ubiquitin ligase to specific substrates. The CBL protein normally exists in the cytosol in an inactive state where the catalytic RF domain is masked by the N-terminal TKB domain. More precisely, the E3 activity tightly depends on a conserved link-helix region (LHR) tyrosine (Tyr371), that in normal conditions, is anchored to the TKB domain and constrains the RING domain in an inactive conformation. When stimulated, CBL is recruited to an activated RTK, leading to extensive tyrosine phosphorylation including Tyr371, which induces conformational changes to activate its E3 activity. This process exposes the RF domain allowing increased E2 binding to the CBL protein in closer proximity to the RTK, facilitating the transfer of ubiquitin from the E2 to lysine residues on the RTK. The ubiquitinated RTK then traffics through the endocytic compartments to the lysosome where it is degraded [11]. CBL and CBL-b share additional areas of homology in the C-terminal half of the proteins, including a ubiquitin-association (UBA) domain and more extensive proline and tyrosine-rich regions, which can be phosphorylated and mediate interactions with Src homology region 2 and 3 (SH2 and SH3) domain-containing proteins including GRB2, CD2AP/CIN85, Cool/Pix and p85 subunit of PI3K [12]. These domains play a critical role in CBL adaptor function. CBL inhibition occurs through dephosphorylation mediated by Src-homology-2-containing phosphatase-1 (SH1P) or ubiquitylation mediated by CBL itself or by the HECT-type E3 ligases atrophin-1-interacting protein-4 (AIP4)/Itch or NEDD4 [13].
3. The Role of CBL in Signaling Pathway Modulation
As previously seen, diverse functions are mediated by the different CBL domains that interact with many protein targets, giving rise to a complex signaling network (Figure 2).
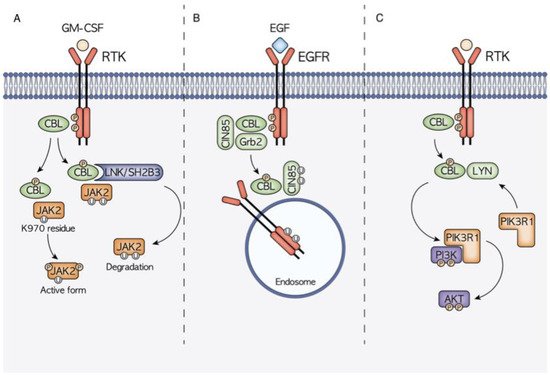
Figure 2. Schematic representation of the three main molecular mechanisms of CBL-mediated signal transduction modulation. (A) CBL is hypothesized to have a dual role in JAK2 signaling. On one hand, thanks to the interaction with the mediator LNK, CBL controls the ubiquitination and consequent degradation of JAK2, on the other hand it has been highlighted how the CBL-mediated ubiquitination at the JAK2 K970 residue causes JAK2 hyperphosphorylation and the consequent activation. (B) CBL is a well-known regulator of EGFR signaling, being able to induce EGFR ubiquitination and degradation. CBL binding can be mediated also by other adaptor proteins such as Grb2 and CIN85. (C) CBL activity also has a high influence on the PI3K/AKT signaling cascade. Through the interaction with LYN, it promotes the recruitment of PIK3R1 and the subsequent PI3K phosphorylation and activation.
The TKB domain that mediates the recognition of phosphorylated tyrosine on activated RTKs, and the RF domain that recruits ubiquitin-conjugating enzymes, are primarily responsible for CBL ubiquitin-protein ligases function that controls ubiquitylation and downregulation of RTKs [13]. CBL has been reported to play a role in different well-known cell pathways, many of which are related to hematopoietic development, immunology and cancer onset and progression. Its double role as an E3 ubiquitin ligase and multi-adaptor protein, which is implicated in positive regulatory functions in signaling transduction, confers a gain of function mechanism in CBL mutant cells, associated with the disruption of E3 ubiquitin ligase activity. The gain of function phenotype is partly lost when there is co-occurrent presence of either the wild-type CBL allele or co-transduction of wild-type CBL. Indeed, this phenotype becomes apparent in the CBL-null background, which supports the observation that CBL mutations are often found in a homozygous state with loss of the wild-type allele. CBL mutations are often missense changes at highly conserved amino-acid positions within the linker and RF domains, which in turn leads to amino acid deletions within these domains. Although the E3 ubiquitin ligase activity primarily depends on the RF domain, the intact linker sequence is also considered structurally essential for an efficient ubiquitinylation to occur [14].
3.1. CBL and JAK2 Signaling
JAK2 is a tyrosine kinase implicated in the signaling of type II cytokine receptor family, including interferon, erythropoietin, granulocyte-macrophage colony-stimulating factor (GM-CSF) and many types of interleukins [15]. Its role is mainly related to the hematopoietic compartment, where it is involved in hematopoiesis and in immune system regulation. JAK2 function is regulated through the control of its phosphorylation status. CBL has been proven to promote GM-CSF-induced JAK2 activation and signaling, promoting its full phosphorylation after CBL-mediated ubiquitination at K970 residue [16]. On the other hand, JAK2 poly-ubiquitination occurs through CBL via the adaptor protein LNK/SH2B3, increasing its proteasomal degradation [17][18][17,18]. The CBL–LNK–JAK2 signaling complex has indeed been proved to regulate JAK2 ubiquitination, stability and activity.
3.2. CBL and EGFR–CBL–CIN85 Axis
CBL is one of the main regulators of the RTK EGFR [10], whose ubiquitination and consequent downregulation are strictly linked to CBL RING and TKB domain functions. The interaction between CBL and EGFR can also be mediated by other interactors such as the adaptor proteins Grb2 and CIN85. CIN85 contains a SH3 domain that is able to bind CBL promoting RTKs trafficking to endosomes prior to their lysosomal degradation [12].
3.3. CBL and PI3K/AKT/LYN Interaction
Another signaling axis playing a significant role in the gain-of-function phenotype conferred by CBL mutations is CBL–LYN–phosphatidylinositol 3-kinase (PI3K). CBL mutations have been reported to increase the expression levels of the PI3K/AKT pathway promoting tumoral onset [19][20][19,20]. This effect has been demonstrated to be strictly linked with the interaction with the LYN protein. In CBL mutant cells, increased LYN activation and interaction with mutant CBL enhance CBL phosphorylation, PI3K regulatory subunit 1 (PIK3R1) recruitment and downstream PI3K/AKT signaling activation [21].
4. CBL in Human Malignancies
4.1. CBL in JMML
The role of CBL as a cancer-predisposing gene was first identified in myeloid neoplasms, particularly in juvenile myelomonocytic leukemia (JMML) [22]. JMML is a rare clonal hematopoietic disorder that typically affects infants and young children, with a median age at presentation of 2 years [23]. It is characterized by alterations in the RAS pathway and about 90% of patients harbor a mutation in one of five genes (PTPN11, NRAS, KRAS, NF1 and CBL) [24]. Clinically, JMML results in anemia, leukocytosis, thrombocytopenia and the infiltration of monocytic and granulocytic cells in different organs, chiefly the spleen.
Homozygous CBL mutations have been found in around 10% to 15% of all JMML cases [25][26][25,26]. Intriguingly, most of these children have a heterozygous germline CBL mutation. Leukemia development is associated with loss of heterozygosity (LOH) occurring, in the majority of cases, via a uniparental disomy resulting in 11q isodisomy in hematopoietic stem cells [27][28][29][30][31][32][27,28,29,30,31,32]. CBL mutations are mainly located throughout the linker and RF domains encoded by exons 8 and 9, especially in correspondence with Y371C [26][28][26,28], and consist of missense mutations or splice site variants [33]. The end result is the expression of a CBL protein with defective E3 ligase activity that constitutively activates the RAS pathway [31]. CBL mutated JMML presents low DNA methylation levels [34] and secondary genetic alterations are not commonly found [29][35][29,35]. The absence of other concomitant RAS pathway-associated mutations may indicate that CBL mutations play a pivotal role in deregulating this key pathway in JMML pathogenesis. Notably, clinical features and methylation profiling are not able to distinguish JMML with CBL germline mutations from patients with only somatic mutations [29].
This subgroup of JMML is associated with a relatively good prognosis. Most patients have a self-limiting indolent clinical course with spontaneous regression of the myeloproliferative disease without any therapy despite the persistence of LOH and clonal hematopoiesis [28][36][37][28,36,37]. Some children instead present an aggressive disease characterized by severe splenomegaly and platelet-transfusion dependency [28][38][39][28,38,39]. Therefore, a strict clinical follow-up without therapeutic intervention is recommended immediately after diagnosis (watch and wait strategy), and allo-HSCT is performed only if chromosomal aberrations occur or in case of disease progression [39]. If necessary, the choice of bridge to transplant therapy remains uncertain [39]. Interestingly, in a recent trial, the hypomethylating agent azacitidine demonstrated to provide a relevant clinical benefit in newly diagnosed JMML prior to HSCT, but patients with CBL mutations were excluded [40].
4.2. CBL in Hematological Neoplasms
Homozygous mutations in CBL have been found in a wide variety of myeloid neoplasms other than JMML. CBL mutations are frequent in chronic myelomonocytic leukemia (5–13% of patients) [41][42][43,44] and acute myeloid leukemia (AML), particularly forms secondary to myelodysplastic syndromes (9% of cases), suggesting that CBL mutations may contribute to leukemia development [9][41][9,43]. Furthermore, CBL mutations could also be found in myelodysplastic syndromes, chronic myelogenous leukemia and other chronic myeloproliferative diseases [9][41][42][9,43,44]. Similar to what has been described in JMML, the majority of patients show missense mutations clustering within the linker region and the RF domain [9]. Less frequently, exon 8 deletions have been reported [9][43][9,45]. Interestingly, most missense mutations are homozygous, while exon 8 deletions are usually heterozygous [9]. Although in vitro studies demonstrated that expression of CBL exon 8 and/or exon 9 deletions in a CBL wild-type context shows a transforming phenotype, in the AML patients this alteration was found only in concomitance with other aberrations, particularly CBF leukemia [44][46]. It is therefore possible to speculate that CBL mutations may cooperate in the pathogenesis of this form of AML [43][44][45,46]. Moreover, the majority of CBL mutations in myeloid neoplasms have been reported in the absence of RAS or PTPN11 mutations, highlighting the mutual exclusivity of CBL mutations and other RAS pathway alterations [9]. The prognostic impact of CBL mutations in myeloid malignancies is still controversial [9][42][45][46][9,44,47,48]. To date, no clear clinical association has been established, and this should be a focus of research in future. Despite somatic mutations in CBL being found in approximately 5% of myeloid neoplasms [9], leukemia other than JMML have not been reported frequently in patients with germline CBL mutations. To date, only two reports exist in the literature of AML developing in patients with CBL syndrome. Becker et al. described a case of AML in an adult patient with a heterozygous de novo germline mutation in CBL codon D390 located in the RF domain [47][49]. Somatic leukemia cells presented with homozygous CBL mutations resulting from copy-neutral LOH and an additional chromosomal gain at 11q. Additional mutations were found in the AML bone marrow, including inv(16) (p13q22) [47][49]. Interestingly, the bone marrow maintained CBL LOH even during complete remission and with normal blood counts, similar to what has been described in JMML [28][36][37][28,36,37]. In another report, Ali et al. addressed the case of an adult patient with missense CBL germline familial mutation in exon 8 developing AML. Leukemia arose due to copy-neutral LOH occurring through uniparental disomy. After induction chemotherapy, morphological remission was achieved, but atypical monocytosis and homozygous CBL mutation persisted in the absence of the previously detected AML-associated mutations and chromosomal alterations. After the consolidation course, the bone marrow still presented the same reported morphological and genetic features. The clinicians therefore suspected an underlying chronic myelomonocytic leukemia or pre-neoplastic monocytic expansion, and performed haploidentical allo-HSCT from her non-CBL-carrying son, achieving complete chimerism and complete remission [48][50]. A homozygous somatic mutation in CBL (p.C381R) associated with an 11q-acquired uniparental disomy was found in one patient with T-ALL but without evidence of germline predisposition [49][51].
4.3. CBL in Other Malignancies
The role of CBL in oncogenesis seems to not be limited to hematological malignancies. Somatic CBL mutations have been reported in lung cancers [50][51][54,55] while diffuse teratomas and embryonal rhabdomyosarcoma have been observed in patients with germline CBL mutations resulting in LOH in the CBL locus [52][53][56,57]. Daniels et al. recently catalogued the mutational spectrum found in various tumors of the three genes: CBL, CBL-b, and CBL-c in the TCGA database [54][58]. The mechanism by which CBL contributes to the pathogenesis of solid tumors has not been completely clear. Other than the already discussed effect on the RAS pathway, a fascinating hypothesis is to consider the role of CBL in tumor-mediated angiogenesis [55][59]. Tumors of c-CBL knockout mice indeed exhibit increased growth and higher angiogenesis compared with wild-type mice [56][60] (Figure 3).
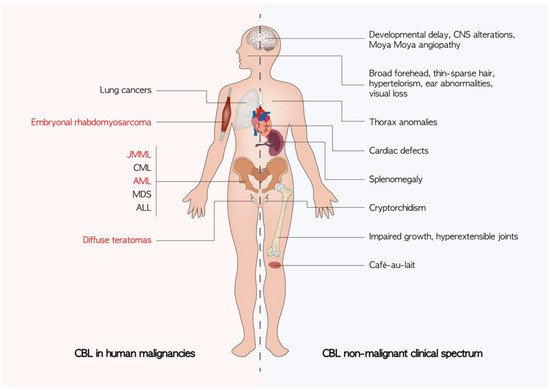
Figure 3. Known clinical manifestations of CBL mutations. The non-malignant phenotypes are an expression of germline CBL mutations. Cancers described in patients harboring a germline CBL mutation are highlighted in red. ALL: acute lymphoblastic leukemia; AML: acute myeloid leukemia; CML: chronic myelomonocytic leukemia; CNS: central nervous system; JMML: juvenile myelomonocytic leukemia; MDS: myelodisplastic syndrome.