Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Adan Pinto-Fernandez and Version 2 by Camila Xu.
Ubiquitylation and ISGylation are protein post-translational modifications (PTMs) and two of the main events involved in the activation of pattern recognition receptor (PRRs) signals allowing the host defense response to viruses.
- SARS-CoV-2
- COVID-19
- ubiquitin proteasome system
1. Introduction
COVID-19 has seen an unprecedented level of research and funding dedicated to fighting the disease. Novel viral vector and nucleic acid vaccines have been developed in record times, expedited by knowledge harbored from preceding outbreaks. Despite the success of global vaccine rollouts, several mutated variants of SARS-CoV-2 have evolved, resulting in subtle changes in disease indications and strengths of transmissibility [1][6]. Due to the likely emergence of future vaccine-resistant variants, it is imperative to develop novel strategies to combat the disease. The post-translational modification of proteins with ubiquitin (ubiquitylation) and/or ISG15 (ISGylation) plays a key role in mediating cellular host–pathogen interactions and antiviral signaling and defense by modulating key events of the innate immune activation signaling.
2. SARS-CoV-2-General Biology and Mechanisms of Infection
2.1. SARS-CoV-2 Viral Genome Architecture
SARS-CoV-2, along with other CoVs, has a genome of approximately 30kB of single-stranded, positive-sense RNA [2][7], making them the largest RNA genomes described to date [3][8]. The virus has 79% sequence identity with SARS-CoV and 50% with MERS-CoV [4][9]. Its genome contains twelve functional open reading frames ordered from 5′–3′ that encode the viral replication transcription complex (ORF1a/1b), the four structural proteins, Spike (S), Envelope (E), Membrane (M), and Nucleocapsid (N), and the likelihood of highly variable accessory proteins encoded throughout [2][7] (Figure 1A). Once viral RNA is released into the cell, it translates ORF 1a/1b to generate the huge replicase polyproteins pp1a and pp1ab, which are responsible for viral transcription, replication, and higher-order RNA structure [2][7]. Sixteen non-structural proteins (nsps) are liberated by the proteolytic cleavage of pp1a (nsp1–11) and pp1ab (nsp1–10, nsp12–16). The enzymes responsible for this are two cysteine proteases; papain-like protease (PLpro) is positioned in the large nsp3 subunit, and the highly conserved main protease (Mpro)/chymotrypsin-like protease (3CLpro) is located within nsp5 (Figure 1B). Mpro cleaves the nsp4–nsp11 region of pp1a and nsp4–nsp16 of pp1ab, whereas PLpro cleaves the nsp1–nsp4 domain [5][10].
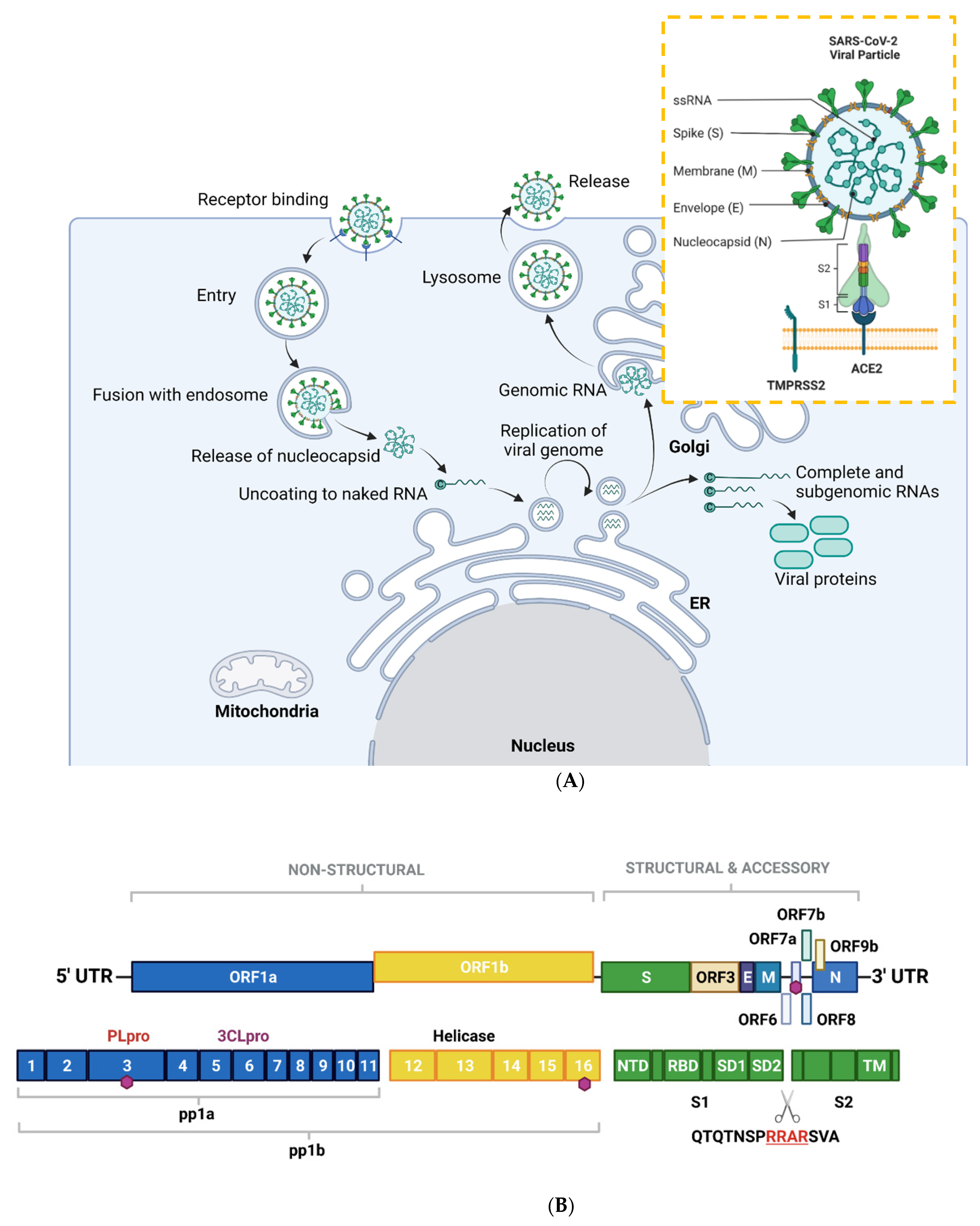
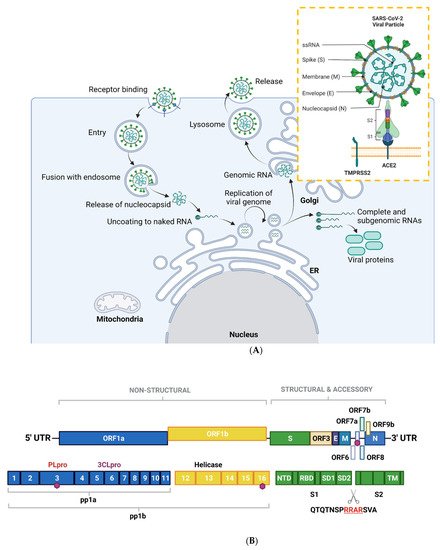
Figure 1. Overview of the SARS-CoV-2 viral life cycle and structural organization. (A) SARS-CoV-2 comprises four structural proteins—the envelope (E) and membrane (M) proteins encase the single-stranded RNA and nucleocapsid (N). The spike (S) protein is made up of S1 and S2 subunits and facilitates receptor binding in the host cell. In humans, this is ACE2, in synergy with TMPRSS2 (orange box). SARS-CoV-2 binds to ACE2 on respiratory epithelial cells through the receptor binding domain on S. The SARS-CoV-2 viral particle is endocytosed following cleavage of S. SARS-CoV-2 nucleocapsid, a complex of viral RNA and N protein, is released into the cytosol. The N protein is removed from the capsid, leaving naked viral RNA, which is a capped positive-stranded RNA molecule that can immediately be translated to allow production of viral proteins (not shown). Viral replication centers are formed in the ER, using the positive-stranded RNA molecule as a template to form a double-stranded RNA intermediate, which in turns allows replication of the viral genome. Complete and sub-genomic RNAs are also formed during the replication process, which serve as the basis for production of a range of viral proteins. The replicated genomic RNA is then bound by N protein to form nucleocapsid, which is encapsulated in vesicles at the Golgi. The contents of the vesicles are then exocytosed, spreading viral particles from the infected cell. (B) Layout of SARS-CoV-2 genome. The virus encodes for ORF1a, ORF1b, ORF3, ORF6, ORF7a/b, ORF8, ORF9b, as well as S, E, M, N. Sixteen non-structural proteins (nsps1–16) of varying function are encoded by OF1a and ORF1b. nsp1 regulates viral mRNAs and interferes with host translation, nsp2–11 facilitate viral replication, nsp12 has RNA polymerase activity, whereas nsp14 is involved in RNA proofreading. A scissors indicates the site of furin and TMPRSS2 cleavage in S1/S2 and S2′, whereas sites of ubiquitin modification are identified by purple hexagons. Other regions of interest include PLpro and 3CLpro. S is composed of several sub-domains including the N-terminal domain (NTD), receptor-binding domain (RBD), subdomains 1 and 2 (SD1, SD2) and the transmembrane domain (TM). Panel B is adapted from Zhang et al., 2021. Figure created with BioRender.com in January 2022.
2.2. SARS-CoV-2 Mode of Entry and Proliferation
An overview of the SARS-CoV-2 viral life cycle is depicted in Figure 1A. As with the original SARS-CoV virus, human SARS-CoV-2 uses angiotensin-converting enzyme 2 (ACE2) as the main receptor for host cell invasion [6][7][11,12]. More recently, CD147 has also been identified as a novel route for infection [8][13]. The heavily glycosylated S protein mediates this attachment to host cell surface receptors [9][5]. Similar to other class I fusion glycoproteins, the viral S protein is cleaved and activated by host cell proteases to form an endosome [10][14]. The protease central to SARS-CoV-2 cell entry is Transmembrane Protease, Serine 2 (TMPSS2; Figure 1A) [11][15]. Once the endosome is formed, SARS-CoV-2 enters the cell either through acidification or the action of host protease cathepsin L.
The S protein is composed of two functional subunits: S1, which is responsible for binding to membrane-bound ACE2 [12][16], and S2, which contains the fusion domain. In the C-terminal to the S1 domain lies the receptor-binding domain (RBD), which is essential for viral entry [7][11][13][12,15,17]. One feature of SARS-CoV-2 that sets it apart from SARS-CoV and other beta-coronaviruses is a four-residue insertion of PRRA, forming a polybasic cleavage site RRAR at the junction of S1 and S2 (Figure 1B). This cleavage site can recruit furin and other host proteases, but it is unclear how this affects general virulence [13][17].
2.3. Clinical Manifestations of COVID-19
SARS-CoV-2 causes a wide spectrum of disease, from asymptomatic illness to severe acute respiratory failure. Findings on autopsy in terminal cases include diffuse alveolar damage, interstitial edema, and reactive type II pneumocytes [14][18]. Since SARS-CoV-2 enters cells via ACE2, there is a gradient of infection across the respiratory tract, with cells in the upper respiratory tract with high ACE2 and TMPRSS2 expression showing higher viral loads [15][19]. It is increasingly recognized that the host immune response is a major factor in the clinical manifestations of COVID-19. Indeed, aspects of the immune system are evaded, whereas others are magnified, leading to profound cytokine release, T cell activation, increased antibodies, and abnormalities of the granulocyte lineage [16][20]. In severe cases, T cells are both depleted and strongly activated [17][21], and peripheral blood has a “cytokine storm” profile, with large concentrations of inflammatory cytokines [16][20]. How SARS-CoV-2 achieves this in vivo has not been fully characterized, but work on airway epithelial cell lines has revealed a complex interaction of delayed, but powerful, type I interferon (IFN) response to SARS-CoV-2 infection, perhaps as a way of gaining time for viral replication [18][22]. Accumulating evidence has demonstrated that numerous IFN-stimulated genes (ISGs) and post-translational ubiquitylation play key roles in cellular antiviral signaling and defense by modulating key events of the innate immune activation signaling. Such mechanisms provide an attractive framework for the development of SARS-CoV-2 therapeutics.
3. Ubiquitylation, ISGylation and Their Roles in Human Antiviral Responses
3.1. Ubiquitylation
The modification of proteins by ubiquitin and ubiquitin-like proteins (Ubls) plays a key role in mediating cellular antiviral signaling and defense. Ubiquitin is an 8.6 kDa protein, and its conjugation to lysine residues on cellular proteins controls many processes, especially targeted protein degradation by the proteasome [19][20][23,24]. There is an extensive network of enzymes that controls the addition and removal of ubiquitin to substrates, which are carried out by the approximately 600 ubiquitin E3 ligases and 100 deubiquitinases (DUBs) in humans [21][25]. The substrate specificity of these enzymes enables varied outcomes of ubiquitin modification. In addition to its N-terminus, ubiquitin itself has seven acceptor lysine (K) sites to which further ubiquitin proteins can be conjugated, allowing the formation of eight basic types polyubiquitin chains. The combinatorial possibilities of ubiquitin chains have led to speculation that their structure can confer signaling information in the cell [22][26]. Recent technologies have begun to reveal an unprecedented landscape of ubiquitin chain composition [23][24][27,28].
3.2. Ubiquitin in Innate Immune Sensing Pathways
Polyubiquitin chains are vital mediators in signaling downstream of three of the main families of innate immune signaling pathways, known as pattern recognition receptor (PRRs): toll-like receptor (TLRs); retinoic acid-inducible gene 1 (RIG)-like receptors (RLRs); and nucleotide-binding oligomerization domain (NOD)-like receptors (NLRs), which act together to sense a variety of Pathogen-Associated Molecular patterns (PAMPs). These pathways all require the assembly of K48- and K63-linked polyubiquitin chains on signaling proteins downstream of PRRs [25][29] (Figure 2). Triggering type I IFN production and release is a crucial outcome of PRR signaling, as type I IFN unleash autocrine and paracrine antiviral responses [26][30]. Research is currently defining the precise role of PRRs in sensing SARS-CoV-2, building on the knowledge of coronavirus infection [27][31]. Therefore, this contributes to the understanding of ubiquitin in antiviral signaling and defense in COVID-19 [28][32].
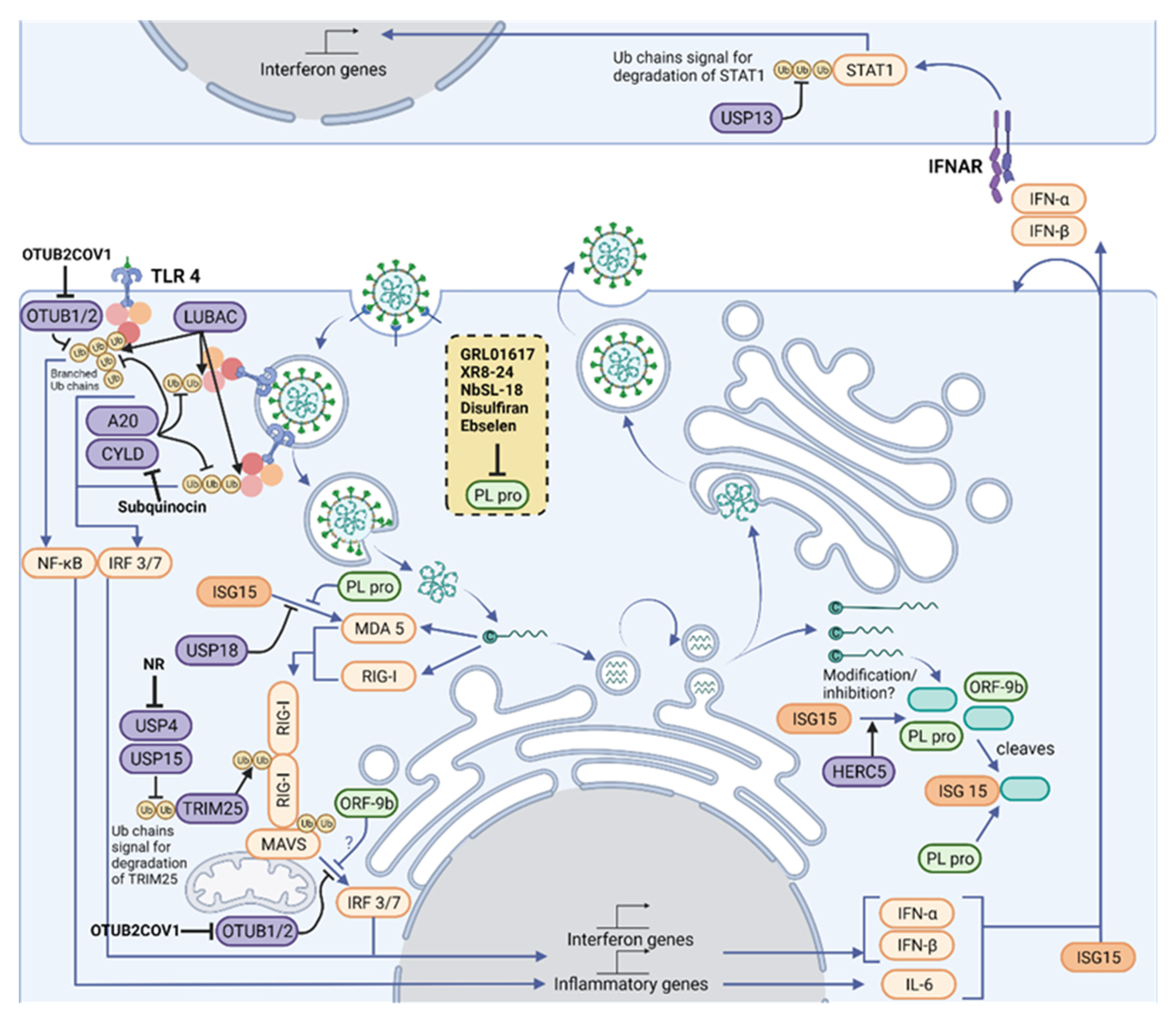
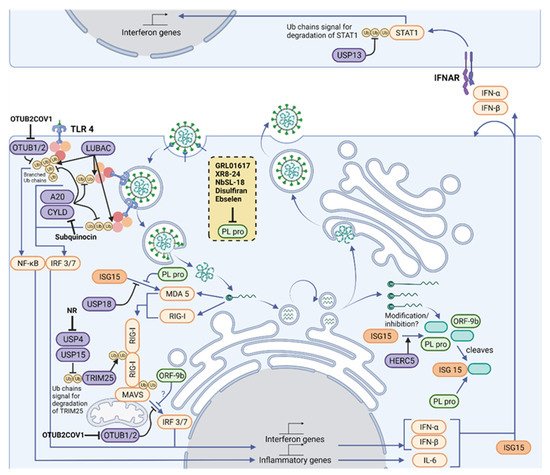
Figure 2. Therapeutic intervention points in the Ubiquitylation and ISGylation machinery of the host and of SARS-CoV-2. Polyubiquitin chains are vital mediators in signaling downstream of three of the main families of innate immune signaling pathways, TLRs, RLRs and NLRs, which act together to sense a variety of PAMPs, including SARS-CoV-2. A network of E3 ligases and DUBs (purple) generate new ubiquitin (yellow) and ISG15 (orange) architectures during viral infection. For instance, LUBAC assembles linear ubiquitin chains on pre-existing K63-linked ubiquitin chains, forming branched structures. Their structures are reshaped and limited by OTUB1/2, OTULIN (not shown), A20 and CYLD. K63-linked ubiquitin chains are assembled on RIG-I by TRIM25, which allow its polymerization and activation of MAVS. In addition, the amount of K48-linked ubiquitin chains regulates TRIM25 levels, and these chains are cleaved by USP4 and USP15. MAVS signaling to IRF3/7 depends on ubiquitylation of NEMO (not shown), but this can be removed by OTUB1/2. Some of these pathways end up activating the IFN-I pathway, driving production of ISGs, including more IFN and ISG15. ISG15 is conjugated by HERC5 to host proteins, which activates antiviral signaling, and to viral proteins, which is believed to inhibit viral processes. Signaling from MDA5 and RIG-I to MAVS depends on the ubiquitylation of MAVS and RIG-I, as well as on the ISGylation of MDA5. Activated MAVS drives IRF3/IRF7-dependent IFN-I gene production. However, the cell also produces USP18, an ISG protease that removes ISG15 from cellular proteins, fine-tuning IFN signals. An antiviral response is driven by neighboring cells through paracrine signaling of type I IFNs. The extent of downstream signaling depends on levels of STAT1, which is modulated by K48-linked ubiquitin chains and its removal by USP13. Host protease inhibitors have been described for CYLD (Subquinocin), OTUB2 (OTUB2COV1) and USP4 (NR). SARS-CoV-2 opposes ubiquitin- and ISG15-mediated antiviral mechanisms by encoding the multi-activity deubiquitylating/deISGylating enzyme PLpro (light green) to inhibit their function. A number of PLpro inhibitors have been identified (listed in the yellow box), characterized and are undergoing clinical trials. Figure created with BioRender.com in January 2022.
The balance of signaling is crucial. Although PRR signaling is key for viral sensing and clearance, an excessive inflammatory response leads to pathological lung inflammation. Proinflammatory cytokines produced downstream of TLR-driven NF-kB signaling pathways are highly elevated in severe cases of COVID-19. In the lung airway space, bronchoalveolar lavage (BAL) washes allows recovery of supernatant for cytokine readouts. These have shown increased Interleukin (IL)-6 and IL-8 production in severe cases COVID-19 [29][33]. Plasma IL-6 is highly elevated, but GM-CSF is suggested to be a particular feature of severe cases of COVID-19, as it not found in patients who have died from influenza virus infection [30][34]. Although immune cells are the most likely cellular source of these cytokines, their precise origin has yet to be elucidated. Understanding which cell types are producing these cytokines will give clues to which viral sensing pathways are hyperactivated in COVID-19.
3.3. TLR Signaling
TLR receptors can detect a variety of viral PAMPs [31][35], and current work is uncovering how SARS-CoV-2 activates TLRs. On first contact with cells, exposed proteins on the surface of SARS-CoV-2 can activate TLR2 and TLR4 (Figure 2). Ubiquitin is important in TLR signaling from surface receptors. Once TLR2 or TLR4 bind a PAMP, the adaptor protein MyD88 is recruited to the intracellular domain of the receptor, where it oligomerizes and forms the basis of a protein complex known as the Myddosome [32][36]. Several members of the IL-1R-associated kinase (IRAK) family are recruited to the Myddosome mediated by death domain interactions of the proteins. The Myddosome platform then recruits E3 ligases TNF receptor-associated factor 6 (TRAF6), Pellino1, and Pellino2, which assemble K63-linked polyubiquitin chains on IRAK1. TRAF6 recruits linear ubiquitin chain assembly complex (LUBAC), which assembles Met1-linked ubiquitin chains. The resulting K63-Met1 hybrid chains serve as a signaling platform that activates the TAK1/TAB2/3 complex and NEMO/IKKα complex, leading to inflammatory gene expression [33][37].
Although TLR4 is mostly known for detecting lipopolysaccharide from Gram negative bacteria, it can bind viral PAMPs such as respiratory syncytial virus (RSV) F protein [34][38], causing proinflammatory signaling. TLR4 is suggested to also sense SARS-CoV-2 S protein [35][39]. In response to S protein, bone-marrow-derived macrophages (BMDMs) from TLR4 knockout mice do not induce the same level of IL-1β mRNA expression as wild-type BMDMs. Further evidence will be needed to confirm this in vivo.
TLR2 has been found to detect the envelope protein of SARS-CoV-2, and induces proinflammatory cytokine production [36][40]. In severe COVID-19, the transcriptome of whole blood shows more mRNA of several TLRs, including TLR2 and the TLR adaptor protein MyD88 in severe COVID-19 [36][40]. This signature could be due to increased numbers of circulating immune cells confounding bulk transcriptomic analysis, rather than upregulation of the mRNAs on an individual cell basis. Nevertheless, this points to an increased capacity for TLR signaling in the blood, suggesting pathological upregulation of TLR signaling in severe COVID-19.
The binding of dsRNA to TLR3 in endosomes results in assembly of a signaling complex including recruitment of ubiquitin ligases such as Pellino1 [37][41] and LUBAC. Pellino1 knockout BMDMs do not upregulate IFN-β production [38][42], and LUBAC deficiency causes impaired TLR3 signaling during influenza virus (IAV) infection [39][43]. Pellino1 has been shown to assemble K63-linked polyubiquitin chains on IRAK-1 in vitro, although this is not yet proven to have role in TLR3 signaling.
The SARS-CoV-2 replication cycle involves a dsRNA intermediate [40][44], which is an activator of TLR3 and TLR7 signaling. The transcription factor interferon regulator factor (IRF) 3 is activated by these pathways, and drives antiviral IFN-I gene expression. In COVID-19, rare genetic variants in TLR3 and TLR7 have been discovered in patients with severe disease, suggesting the importance of these pathways [41][42][45,46]. TLR3 knockout mice are more susceptible to SARS-CoV infection [43][47], and TLR3/7 knockdown impairs SARS-CoV-2 sensing of Calu-3 cells [44][48].
3.4. RLR Signaling
Coronaviruses have previously been identified as activators of RLR signaling in murine models [45][46][49,50]. RLR signaling detects viral RNA and depends on assembly of polyubiquitin chains for signaling [47][51]. There are three sensors in the RLR family, all of which are cytosolic: RIG-I and MDA5 initiate a signaling pathway that causes production of type I IFNs, whereas LGP2 regulates RIG-I signaling. When RIG-I is inactive, the Caspase Activation and Recruitment Domains (CARD) are sequestered in the protein. However, on binding RNA molecules with a 5′ triphosphate group, the CARD is released and mediates downstream signaling with Mitochondrial Anti-Viral-Signaling protein (MAVS). Less is known about the mechanism of MDA5 activation, with single- or double-stranded RNA proposed as a ligand.
RIG-I activation depends on a carefully tuned network of E3 ligases and DUBs to promote signaling and balance degradation. The CARD domain opening in RIG-I depends on K63-linked polyubiquitylation. A structure of RIG-I-Ub2 (diubiquitin) has shown the mechanism of how polyubiquitylation mediates the signaling. Non-covalent interactions between the ubiquitin and CARDs enable tetramerization of the CARD subunits, generating a binding surface that can activate a CARD on MAVS, allowing downstream signaling [48][52]. The ubiquitylation of RIG-I is controlled by several E3 ligases, including TRIM25 and TRIM4 [49][50][53,54], with redundancy likely being important due to selective pressure from viruses. Signaling is inhibited by the DUBs CYLD and USP3, which remove the K63-linked chains [51][52][55,56]. In addition to controlling signaling, ubiquitylation also regulates protein levels through K48-linked ubiquitin chains in RLR signaling. RIG-I can be ubiquitinated with K48-linked chains by Riplet [53][57]. RIG-I and TRIM25 can be stabilized through removal of K48-linked chains by USP4 and USP15, promoting signaling [54][55][58,59].
The role of ubiquitin in MDA5 signaling is less clear. MDA5 is activated by K63-linked ubiquitylation of its helicase domain [56][60], and its CARDs have a lower affinity for K63-linked ubiquitin, suggesting a different mechanism than RIG-I activation. In SARS-CoV-2 infection, MDA5 activation has also been found to depend on modification with a ubiquitin-like protein (Ubl), ISG15 [57][61], which will be expanded in the section on Ubls in antiviral defense.
The mechanism of SARS-CoV-2 sensing by RLRs is currently being elucidated. Most studies so far have looked at RLR signaling in Calu-3 cells, a lung adenocarcinoma cell line that expresses the factors required for SARS-CoV-2 entry, TMPRSS2 and ACE2 [58][59][60][61][62,63,64,65]. Thesere papers all show the importance of MAVS and thus RLR signaling in the detection of SARS-CoV-2. Three of these studies suggest that MDA5 is the key sensor and RIG-I is dispensable [59][60][62][63,64,66], but Thorne et al. suggest that RIG-I depletion does affect SARS-CoV-2 sensing [60][64]. A study of primary bronchial epithelial and alveolar cell infection by SARS-CoV-2 suggest the opposite, that RIG-I sensing is required whereas MDA5 is dispensable [62][66]. More studies will be required in different cell lines and in vivo research to clarify whether RIG-I is contributing to SARS-CoV-2 sensing.
Once activated by RIG-I or MDA5, MAVS forms filaments on the surface of the mitochondria [47][51]. This complex activates the transcription factor dimer IRF3/7, which drive expression of IFNs and ISGs. MAVS is also ubiquitylated by TRIM25, however in this case, TRIM25 assembles K48-linked chains on MAVS which cause its proteasomal degradation. MAVS degradation is required to release a signaling complex including NEMO and TBK1 [63][67]. K63-linked ubiquitin chains on NEMO serve as a platform to activate the kinase TBK1 and allow phosphorylation of IRF3. SARS-CoV-2 can modulate NEMO ubiquitylation to oppose MAVS signaling, which will be explored in the section on the viral modulation of ubiquitin signaling [64][68].