Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Catherine Yang and Version 1 by Paweł Kowalczyk.
The excessive formation of reactive oxygen species (ROS) and impairment of defensive antioxidant systems leads to a condition known as oxidative stress. The main source of free radicals responsible for oxidative stress is mitochondrial respiration. The deleterious effects of ROS on cellular biomolecules, including DNA, is a well-known phenomenon that can disrupt mitochondrial function and contribute to cellular damage and death, and the subsequent development of various disease processes.
- mitochondrial diseases
- oxidative stress
1. Neurological Diseases
Neurodegenerative diseases such as Alzheimer’s disease (AD), Parkinson’s disease (PD), and amyotrophic lateral sclerosis (ALS) are age-related conditions characterized by significant changes in mitochondrial structure and function associated with free radicals generation [36][1].
Increased levels of free radicals and higher oxidation of macromolecules including mtDNA have been observed in Alzheimer’s disease (AD) human brains and in various animal models [37][2]. What is more, free radicals have been shown to increase the activity of β- and γ-secretases, enzymes responsible for amyloid β generation from amyloid precursor protein [38][3]. Further, Nonomura et al. [39][4] demonstrated that oxidative damage is quantitatively greatest early in the AD and decreases with dementia progression and amyloid β plaque deposition. It has been proposed that mitochondrial oxidative stress damages mtDNA encoding electron transfer chain subunits, which negatively affects ATP production and calcium homeostasis, and exacerbates oxidative stress. The latter, in turn, increases amyloid β deposition and leads to further consequences of neuronal dysfunction, neurodegeneration, and cognitive impairment in AD [36,40,41,42][1][5][6][7].
It is still not clear whether mitochondrial dysfunction plays a direct role in the initiation of AD according to the “mitochondrial cascade hypothesis” or is, rather, a consequence of amyloid β accumulation. Indeed, Reddy [43][8] suggested that progressive mitochondrial damage leading to disease progression is caused by β-amyloid entry into mitochondria, triggering the production of free radicals. Oxidative damage and neuroinflammation have been shown to correlate with Alzheimer’s disease progression [41,44][6][9]. A synergistic role of both pathways is also possible [41,45][6][10]. It is certain, however, that many of the therapies targeting mitochondrial dysfunction in neurodegeneration and cognitive dysfunction in AD rely on the application of antioxidants and a reduction in free radical levels [42][7].
Mitochondrial damage closely related to oxidative stress seems to play an important role in the pathogenesis of Parkinson’s disease (PD) [36,46,47][1][11][12]. At the cellular level, PD is caused by both the overproduction of reactive oxygen species and changes in dopamine metabolism, as well as alteration in the mitochondrial electron transporter chain function in the neurons of substantia nigra [48][13]. The involvement of oxidative stress in dopaminergic cell degeneration was indicated further by the increased oxidative damage to mtDNA noted in PD neurons of substantia nigra [49,50,51][14][15][16]. Even mutations in genes coding proteins linked to PD such as DJ-1, parkin, PINK1, alpha-synuclein, and LRRK2 affect mitochondrial function and integrity, causing enhanced ROS generation and vulnerability to oxidative stress [48,52][13][17]. Currently, the role of antioxidant neurotrophic strategies in PD treatment is emphasized. One of them is the proposal to combine antioxidant therapy with stem cell therapy to reduce damage and induce repair of dopaminergic neurons for the treatment of Parkinson’s disease [48,53][13][18].
Oxidative stress exacerbating damage to mitochondria has been also identified as one of the factors involved in demyelination, axonal and neuronal death in multiply sclerosis (MS), and motoneuron death in amyotrophic lateral sclerosis (ALS) [54,55,56][19][20][21]. Undoubtedly, an inflammatory process engaged in oligodendrocyte pathology that activates and recruits lymphocytes, macrophages, and microglia is able to generate vast quantities of oxidizing radicals contributing to MS tissue injury [57][22]. In the case of ALS pathology, the involvement of ROS is supported by the elevated free radical levels in the cerebrospinal fluid, serum, and urine of patients with sporadic and familial forms of ALS [55,58,59][20][23][24]. In addition, in familial ALS, altered reactivity of superoxide dismutase, responsible for the clearance of reactive oxygen species, is reported [60][25]. As shown by Petrozziello et al. [61][26], oxidative stress in ALS causes mitochondrial fragmentation and dysfunction. Unfortunately, clinical trials of antioxidant therapy appear to be unsuccessful despite beneficial effects in animal models [62][27]. Recently, the reduction of oxidative stress damage has been shown to effectively prolong animal survival time and reduce brain pathological symptoms in a mouse model of ALS [62,63][27][28].
The causes of schizophrenia are as yet undetermined. One hypothesis points to oxidative stress as the contributing factor to the pathophysiology of the disease [1,64][29][30]. This is supported by decreased levels of antioxidants and augmented oxidative stress markers in schizophrenic patients [65,66,67,68][31][32][33][34]. Significantly reduced glutathione (antioxidant) levels have been reported in magnetic resonance spectroscopy in the cerebral cortexes of living patients [69][35], but also in post-mortem examination [70][36]. Computer tomography scans showing brain atrophy in chronic schizophrenic patients revealed strong correlation between brain pathology and low glutathione peroxidase activity in platelets [1,71][29][37]. In addition, the oxidative imbalance in schizophrenia was paralleled by increased severity of negative symptoms of the disease [66][32]. Cuenod and colleagues [72][38] emphasize the role of complex mechanisms of oxidative stress and its modulation in the pathophysiology of schizophrenia, and attribute a major role to dysregulation of redox mechanisms, disruption of mitochondrial bioenergetics, and neuroinflammation in the development of oxidative stress during neurodevelopment. The role of one of the forms of oxidative stress, the so-called carbonyl stress, is currently being studied in the pathophysiology of schizophrenia. Hara et al. [73][39] indicate that this stress causes mitochondrial damage, lowers mitochondrial membrane potential, and hinders aerobic respiration processes. Even genetic predisposition linked to mitochondrial function and subsequent oxidative stress has been found; gene cacna1c is considered as a strong genetic risk factor for the development of affective disorders [74][40]. Although the evidence is inconsistent, there are studies demonstrating the efficacy of antioxidant therapies in the treatment of schizophrenia that support the hypothesis that oxidative stress plays an important role in its development [64][30].
2. Neurodevelopmental Disorders
Oxidative stress induced by prenatal exposure to toxic chemicals is regarded as a key factor in the occurrence of neurodevelopmental disorders [75][41]. In the case of autism mitochondrial abnormality, augmented oxidative stress and decreased antioxidant capacity have been reported in autistic persons, all of which may be responsible for neuroinflammation and autism pathology [76,77][42][43]. Recent analysis of blood samples from children with autism spectrum disorders revealed reduced total plasma peroxidase and total antioxidant capacity, resulting in an imbalance in the oxidant/antioxidant ratio and abnormalities in neuronal transduction [78][44]. Zawadzka et al. [79][45] showed that impaired brain development is a consequence of inflammatory processes inducing oxidative stress and mitochondrial damage, which in turn exacerbate oxidative stress, triggering further cellular damage. In support of the role of oxidative stress in autism pathology, studies using n-acetylcysteine or other antioxidants have reported a reduction in some autistic behaviors in children, such as irritability and hyperactivity [76,80,81][42][46][47].
3. Autoimmune Diseases
Another group of diseases whose pathomechanism may involve mitochondrial dysfunction causing oxidative stress are T cell-mediated autoimmune diseases such as type 1 diabetes (T1D), multiple sclerosis (MS), rheumatoid arthritis (RA), and systemic lupus erythematosus (SLE) [82,83][48][49]. The autoreactive T cells that recognize systemic or organ-specific self-antigens, responsible for autoimmunity, are susceptible to ROS that are engaged in their differentiation, effector responses, and inducing proinflammatory cytokine release [82,84][48][50]. The latter triggers inflammation involved in the pathomechanism of autoimmune disorders, resulting in oxidative stress and damage to cellular macromolecules. Oxidative stress and inflammation are closely related. Mitochondrial-derived ROS via the oxidation of biomolecules or structural modification of proteins and genes may start signaling cascades, leading to inflammatory processes. ROS-activated transcription factors and pro-inflammatory genes induce inflammation and recruitment of immune and inflammatory cells to the site of oxidative stress. Activated immune cells generate ROS at the site of inflammation, amplifying oxidative stress and tissue injury [5,85,86,87][51][52][53][54].
In SLE, patients show increased ROS in T cells as well as more oxidized lipoproteins, which can lead to vascular inflammation and atherosclerosis [88][55]. Another pathway of action of ROS on the development of an autoimmune SLA is the damage of DNA, which becomes a major antigenic target for autoantibodies [89][56].
In T1D, profound metabolic changes occur during insulin deprivation including an increase in basal energy expenditure and reduced mitochondrial function [89,90][56][57]. Sustained hyperglycemia induces increased ROS production, and systemic oxidative stress has been confirmed at early onset of T1D, as well as its increase in early adulthood [89,91][56][58]. Indeed, mitochondria-derived free radicals has been demonstrated to contribute to the process of immune-mediated beta-cell destruction via the induction of cytokine toxicity in T1D [89,92][56][59]. Another reason is that beta-cells exhibit insufficient antioxidant defense, which is associated with low expression of antioxidant enzymes in islets [84][50].
The chronic oxidative stress in the RA is characterized by a significant increase in mitochondrial ROS production [93][60]. It contributes to joint damage, playing the role of messenger in inflammatory and immunological cellular response including activation of the NLRP3 inflammasome, which produces cytokines linked to RA symptoms [83][49].
4. Kidney and Lung Diseases
Other diseases associated with mitochondrial oxidative stress and inflammation are chronic kidney disease (CKD) and chronic obstructive pulmonary disease (COPD). Mitochondrial dysfunction, such as decreased mtDNA, and ATP production, as well as the loss of mitochondrial membrane potential, related to increased mitochondrial ROS, has been shown to precede kidney injury and further contribute to the development and progression of CKD, characterized by a decrease in the number of active nephrons [94][61]. Excess ROS present early during CKD progression and contribute to inflammatory process in the renal parenchyma via inflammatory cell recruitment and proinflammatory cytokine production, leading to endothelial impairment and atherosclerosis [95][62]. Interestingly, the mechanism of nephrotoxicity of some drugs (cyclosporine, gentamycin) has been demonstrated to involve oxidative stress induction and lipid peroxidation [96][63].
A leading cause of COPD is cigarette smoking. Cigarette smoke, particulate matter, and noxious gases including ozone are major exogenous sources of ROS that challenge respiratory epithelial cells and injure small airways and lung parenchyma directly or indirectly by increasing inflammation [97,98,99][64][65][66]. Nevertheless, inflammation and oxidative stress are inextricably linked. Indeed, oxidative stress-induced tissue damage can trigger inflammation and immune responses, which in turn can enhance ROS production [5,100][51][67].
Airway smooth muscle and bronchial biopsies from COPD patients showed increased mtROS production and decreased antioxidant enzymes compared to healthy control subjects [101,102][68][69]. Further, impaired redox regulation associated with cellular ageing has been described to contribute to the development and acceleration of COPD pathogenesis via enhanced inflammation, protease–anti-protease imbalance, and cellular apoptosis [103][70].
5. Cardiovascular Diseases (CVDs)
ROS are considered as one of the major causative factors leading to atherosclerosis development. Oxidative stress contributes to atherosclerotic plaque formation via induction of endothelial dysfunction, vascular inflammation, and accumulation of oxidized low-density lipoprotein [104][71]. All these lead to lesion formation and accumulation of macrophages, which, apart from producing ROS, phagocytize oxidized lipoproteins and transform into foam cells, components of atherosclerotic plaque [105,106][72][73]. Oxidative stress markers have been shown to be elevated in patients suffering from cardiovascular diseases such as hypertension [107,108][74][75] and heart failure, whereas its increase in cardiomyocytes is correlated with the development and the progression of maladaptive myocardial remodeling [109,110,111][76][77][78]. Cardiac dysfunction associated with metabolic syndrome comprising of diabetes, high blood pressure, and obesity is actually due to enhanced oxidative stress causing damage of mitochondria, the activation of mitochondria apoptotic signaling pathways, and cardiomyocyte contractile dysfunction [112][79].
6. Cancer
Elevated ROS mutagenicity results from the induction of genetic instability evoked via increasing receptor and oncogene activity, stimulation of oxidative enzymes or growth factor-signaling pathways involved in regulation of DNA repair, cell proliferation, apoptosis, and tumorigenesis [114,115,116][81][82][83].
As mentioned earlier, excess ROS can also directly damage DNA by causing single- and double-strand nucleic acid breaks and by forming an oxidized derivative of deoxyguanosine, 8-Oxo-2′-deoxyguanosine, which contribute to carcinogenesis through promoting mutagenesis [116][83]. Consequently, mutations in mtDNA, reduced mtDNA content, and mutations in nuclear genes can irreversibly damage mitochondrial oxidative phosphorylation. The latter leads to mitochondrial dysfunction and further genetic instability in the nuclear genome, and is one of the proposed causes of cancer [116,117][83][84].
Not surprisingly, oxidative stress may be responsible for the onset and development of various types of cancer from hepatocellular carcinoma, breast cancer, and lung cancer to brain tumors [118,119,120][85][86][87]. ROS have been also shown to induce DNA hypermethylation, which can affect the tumor phenotype [114,120][81][87].
Oxidative stress can act on cancer cells in two ways, which should be taken into account in the design of anti-cancer drugs targeting ROS. In physiological amounts, ROS contribute to further cancer growth by transducing signals for cell proliferation, migration, and angiogenesis, whereas severe oxidative stress may produce a deleterious effect through the induction of cell-cycle arrest and apoptosis [116][83]. However, cancer cells are able to resist excessive intracellular ROS by activating the transcription factor and nuclear erythroid 2-related factor (NRF2) responsible for antioxidant enzymes transcription, promoting cancer cell survival [116,120][83][87].
All disease entities induced by mitochondrial damage are presented in Figure 1.
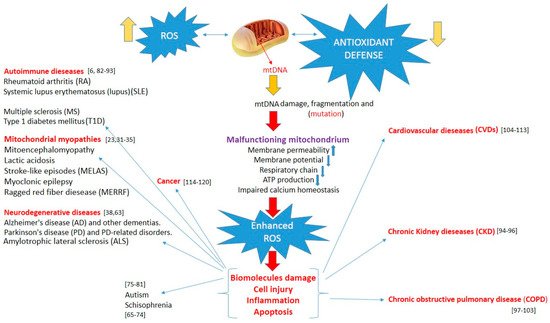
Figure 1. Increased reactive oxygen species, overwhelming antioxidant defenses, induce mtDNA damage, and mitochondrial dysfunction lead to enhanced oxidative stress. This, in turn, can induce biomolecule and cell damage, apoptosis, and inflammation, triggering various pathologies.
References
- D’Errico, M.; Parlanti, E.; Pascucci, B.; Filomeni, G.; Mastroberardino, P.G.; Dogliotti, E. The interplay between mitochondrial functionality and genome integrity in the prevention of human neurologic diseases. Arch. Biochem. Biophys. 2021, 23, 108977.
- Mancuso, M.; Orsucci, D.; Siciliano, G.; Murri, L. Mitochondria, mitochondrial DNA and Alzheimer’s disease. What comes first? Curr. Alzheimer Res. 2008, 5, 457–468.
- Butterfield, D.A. β-amyloid-associated free radical oxidative stress and neurotoxicity: Implications for Alzheimer’s disease. Chem. Res. Toxicol. 1997, 10, 495–506.
- Nunomura, A.; Perry, G.; Aliev, G.; Hirai, K.; Takeda, A.; Balraj, E.K.; Jones, P.K.; Ghanbari, H.; Wataya, T.; Shimohama, S.; et al. Oxidative damage is the earliest event in Alzheimer disease. J. Neuropathol. Exp. Neurol. 2001, 60, 759–767.
- Yakes, F.M.; Van Houten, B. Mitochondrial DNA damage is more extensive and persists longer than nuclear DNA damage in human cells following oxidative stress. Proc. Natl. Acad. Sci. USA 1997, 94, 514–519.
- Swerdlow, R.H. Mitochondria and Mitochondrial Cascades in Alzheimer’s Disease. J. Alzheimers Dis. 2018, 62, 1403–1416.
- Sharma, C.; Kim, S.; Nam, Y.; Jung, U.J.; Kim, S.R. Mitochondrial Dysfunction as a Driver of Cognitive Impairment in Alzheimer’s Disease. Int. J. Mol. Sci. 2021, 22, 4850.
- Reddy, P.H. Amyloid precursor protein-mediated free radicals and oxidative damage: Implications for the development and progression of Alzheimer’s disease. J. Neurochem. 2006, 96, 1–13.
- Li, H.; Knight, W.C.; Xu, J. Striatal oxidative damages and neuroinflammation correlate with progression and survival of Lewy body and Alzheimer diseases. Neural Regen. Res. 2022, 17, 867–874.
- Caito, S.W.; Aschner, M. Mitochondrial redox dysfunction and environmental exposures. Antioxid. Redox Signal. 2015, 23, 578–595.
- Davie, C.A. A review of Parkinson’s disease. Br. Med. Bull. 2008, 80, 109–127.
- Michel, P.P.; Hirsch, E.C.; Hunot, S. Understanding Dopaminergic Cell Death Pathways in Parkinson Disease. Neuron 2016, 90, 675–691.
- Blesa, J.; Trigo-Damas, I.; Quiroga-Varela, A.; Jackson-Lewis, V.R. Oxidative stress and Parkinson’s disease. Front. Neuroanat. 2015, 9, 91.
- Pyle, A.; Anugrha, H.; Kurzawa-Akanbi, M.; Yarnall, A.; Burn, D.; Hudson, G. Reduced mitochondrial DNA copy number is a biomarker of Parkinson’s disease. Neurobiol. Aging 2016, 38, e7–e216.
- Grünewald, A.; Rygiel, K.A.; Hepplewhite, P.D.; Morris, C.M.; Picardm, M.; Turnbullm, D.M. Mitochondrial DNA Depletion in Respiratory Chain-Deficient Parkinson Disease Neurons. Ann. Neurol. 2016, 79, 366–378.
- Dölle, C.; Flønes, I.; Nido, G.S.; Miletic, H.; Osuagwu, N.; Kristoffersen, S.; Lilleng, P.K.; Larsen, J.P.; Tysnes, O.B.; Haugarvoll, K.; et al. Defective mitochondrial DNA homeostasis in the substantia nigra in Parkinson disease. Nat. Commun. 2016, 22, 13548.
- Dias, V.; Junn, E.; Mouradian, M.M. The Role of Oxidative Stress in Parkinson’s Disease. J. Parkinsons Dis. 2013, 3, 461–491.
- Silva, R.; Domingues, H.S.; Salgado, A.J.; Teixeira, F.G. From regenerative strategies to pharmacological approaches: Can we fine-tune treatment for Parkinson’s disease? Neural Regen. Res. 2022, 17, 933–936.
- Adamczyk, B.; Adamczyk-Sowa, M. New Insights into the Role of Oxidative Stress Mechanisms in the Pathophysiology and Treatment of Multiple Sclerosis. Oxid. Med. Cell. Longev. 2016, 2016, 1973834.
- Smith, E.F.; Shaw, P.J.; De Vos, K.J. The role of mitochondria in amyotrophic lateral sclerosis. Neurosci. Lett. 2019, 710, 132933.
- Obrador, E.; Salvador-Palmer, R.; López-Blanch, R.; Jihad-Jebbar, A.; Vallés, S.L.; Estrela, J.M. The Link between Oxidative Stress, Redox Status, Bioenergetics and Mitochondria in the Pathophysiology of ALS. Int. J. Mol. Sci. 2021, 22, 6352.
- Ohl, K.; Tenbrock, K.; Kipp, M. Oxidative stress in multiple sclerosis: Central and peripheral mode of action. Exp. Neurol. 2016, 277, 58–67.
- Simpson, E.P.; Henry, Y.K.; Henkel, J.S.; Smith, R.G.; Appel, S.H. Increased lipid peroxidation in sera of ALS patients: A potential biomarker of disease burden. Neurology 2004, 62, 1758–1765.
- Nissanka, N.; Moraes, C.T. Mitochondrial DNA damage and reactive oxygen species in neurodegenerative disease. FEBS Lett. 2018, 592, 728–742.
- Wiedau-Pazos, M.; Goto, J.J.; Rabizadeh, S.; Gralla, E.B.; Roe, J.A.; Lee, M.K.; Valentine, J.S.; Bredesen, D.E. Altered reactivity of superoxide dismutase in familial amyotrophic lateral sclerosis. Science 1996, 271, 515–518.
- Petrozziello, T.; Bordt, E.A.; Mills, A.N.; Kim, S.E.; Sapp, E.A.; Devlin, B.A.; Obeng-Marnu, A.A.; Farhan, S.M.K.; Amaral, A.C.; Dujardin, S.; et al. Targeting Tau Mitigates Mitochondrial Fragmentation and Oxidative Stress in Amyotrophic Lateral Sclerosis. Mol. Neurobiol. 2021, 10.
- Carrera-Juliá, S.; Moreno, M.L.; Barrios, C.; de la Rubia Ortí, J.E.; Drehmer, E. Antioxidant alternatives in the treatment of amyotrophic lateral sclerosis: A comprehensive review. Front. Physiol. 2020, 11, 63.
- Sugimoto, K.; Liu, J.; Li, M.; Song, Y.; Zhang, C.; Zhai, Z.; Gao, Y. Neuroprotective Effects of Shenqi Fuzheng Injection in a Transgenic SOD1-G93A Mouse Model of Amyotrophic Lateral Sclerosis. Front. Pharmacol. 2021, 19, 701886.
- Wu, J.Q.; Kosten, T.R.; Zhang, X.Y. Free radicals, antioxidant defense systems, and schizophrenia. Prog. Neuropsychopharmacol. Biol. Psychiatry 2013, 46, 200–206.
- Murray, A.J.; Rogers, J.C.; Katshu, M.Z.U.H.; Liddle, P.F.; Upthegrove, R. Oxidative Stress and the Pathophysiology and Symptom Profile of Schizophrenia Spectrum Disorders. Front. Psychiatry 2021, 12, 703452.
- Raffa, M.; Atig, F.; Mhalla, A.; Kerkeni, A.; Mechri, A. Decreased glutathione levels and impaired antioxidant enzyme activities in drug-naive first-episode schizophrenic patients. BMC Psychiatry 2011, 11, 124.
- Gunes, M.; Altindag, A.; Bulut, M.; Demir, S.; Ibiloglu, A.O.; Kaya, M.C.; Atli, A.; Aksoy, N. Oxidative metabolism may be associated with negative symptoms in schizophrenia. Psychiatry Clin. Psychopharmacol. 2017, 27, 54–61.
- Solberg, D.K.; Refsum, H.; Andreassen, O.A.; Bentsen, H. A five-year follow-up study of antioxidants, oxidative stress and polyunsaturated fatty acids in schizophrenia. Acta Neuropsychiatr. 2019, 31, 202–212.
- Dietrich-Muszalska, A.; Kwiatkowska, A. Generation of superoxide anion radicals and platelet glutathione peroxidase activity in patients with schizophrenia. Neuropsych. Dis. Treat. 2014, 10, 703–709.
- Do, K.Q.; Trabesinger, A.H.; Kirsten-Kruger, M.; Lauer, C.J.; Dydak, U.; Hell, D.; Holsboer, F.; Boesiger, P.; Cuénod, M. Schizophrenia: Glutathione deficit in cerebrospinal fluid and prefrontal cortex in vivo. Eur. J. Neurosci. 2000, 12, 3721–3728.
- Gawryluk, J.W.; Wang, J.F.; Andreazza, A.C.; Shao, L.; Young, L.T. Decreased levels of glutathione, the major brain antioxidant, in post-mortem prefrontal cortex from patients with psychiatric disorders. Int. J. Neuropsychopharmacol. 2011, 14, 123–130.
- Yao, J.K.; Keshavan, M.S. Antioxidants, redox signaling, and pathophysiology in schizophrenia: An integrative view. Antioxid. Redox Signal. 2011, 15, 2011–2035.
- Cuenod, M.; Steullet, P.; Cabungcal, J.H.; Dwir, D.; Khadimallah, I.; Klauser, P.; Conus, P.; Do, K.Q. Caught in vicious circles: A perspective on dynamic feed-forward loops driving oxidative stress in schizophrenia. Mol. Psychiatry 2021, 1–12.
- Hara, T.; Toyoshima, M.; Hisano, Y.; Balan, S.; Iwayama, Y.; Aono, H.; Futamura, Y.; Osada, H.; Owada, Y.; Yoshikawa, T. Glyoxalase I disruption and external carbonyl stress impair mitochondrial function in human induced pluripotent stem cells and derived neurons. Transl. Psychiatry 2021, 11.
- Michels, S.; Wöhr, M.; Schwarting, R.K.; Culmsee, C. Psychiatric risk gene cacna1c determines mitochondrial resilience against oxidative stress in neurons. Cell Death Dis. 2018, 9, 645.
- Nishimura, Y.; Kanda, Y.; Sone, H.; Aoyama, H. Oxidative Stress as a Common Key Event in Developmental Neurotoxicity. Oxid. Med. Cell. Longev. 2021, 19, 6685204.
- Balachandar, V.; Rajagopalan, K.; Jayaramayya, K.; Jeevanandam, M.; Iyer, M. Mitochondrial dysfunction: A hidden trigger of autism? Genes Dis. 2020, 8, 629–639.
- Toscano, C.V.A.; Barros, L.; Lima, A.B.; Nunes, T.; Carvalho, H.M.; Gaspar, J.M. Neuroinflammation in autism spectrum disorders: Exercise as a “pharmacological” tool. Neurosci. Biobehav. Rev. 2021, 129, 63–74.
- Omotosho, I.O.; Akinade, A.O.; Lagunju, I.A.; Yakubu, M.A. Oxidative stress indices in ASD children in Sub-Sahara Africa. J. Neurodev. Disord. 2021, 13, 50.
- Zawadzka, A.; Cieślik, M.; Adamczyk, A. The Role of Maternal Immune Activation in the Pathogenesis of Autism: A Review of the Evidence, Proposed Mechanisms and Implications for Treatment. Int. J. Mol. Sci. 2021, 22, 11516.
- Hardan, A.Y.; Fung, L.K.; Libove, R.A.; Obukhanych, T.V.; Nair, S.; Herzenberg, L.A.; Frazier, T.W.; Tirouvanziam, R. A randomized controlled pilot trial of oral N-acetylcysteine in children with autism. Biol. Psychiatry 2012, 71, 956–961.
- Liu, Y.; Yang, Z.; Du, Y.; Shi, S.; Cheng, Y. Antioxidant interventions in autism spectrum disorders: A meta-analysis. Prog. Neuropsychopharmacol. Biol. Psychiatry 2021, 113, 110476.
- Chavez, M.D.; Tse, H.M. Targeting Mitochondrial-Derived Reactive Oxygen Species in T Cell Mediated Autoimmune Diseases. Front. Immunol. 2021, 12, 703972.
- Clayton, S.A.; MacDonald, L.; Kurowska-Stolarska, M.; Clark, A.R. Mitochondria as Key Players in the Pathogenesis and Treatment of Rheumatoid Arthritis. Front. Immunol. 2021, 12, 673916.
- Chen, J.; Stimpson, S.E.; Fernandez-Bueno, G.A.; Mathews, C.E. Mitochondrial Reactive Oxygen Species and Type 1 Diabetes. Antioxid. Redox Signal. 2018, 29, 1361–1372.
- Hussain, T.; Tan, B.; Yin, Y.; Blachier, F.; Tossou, M.C.; Rahu, N. Oxidative Stress and Inflammation: What Polyphenols Can Do for Us? Oxid. Med. Cell. Longev. 2016, 2016, 7432797.
- Chatterjee, S. Chapter Two—Oxidative Stress, Inflammation, and Disease. In Oxidative Stress and Biomaterials; Dziubla, T., Butterfield, D.A., Eds.; Academic Press: Cambridge, MA, USA, 2016; pp. 35–58.
- Singh, V.; Ubaid, S. Role of Silent Information Regulator 1 (SIRT1) in Regulating Oxidative Stress and Inflammation. Inflammation 2020, 43, 1589–1598.
- Geto, Z.; Molla, M.D.; Challa, F.; Belay, Y.; Getahun, T. Mitochondrial Dynamic Dysfunction as a Main Triggering Factor for Inflammation Associated Chronic Non-Communicable Diseases. J. Inflamm. Res. 2020, 13, 97–107.
- Wincup, C.; Radziszewska, A. Abnormal Mitochondrial Physiology in the Pathogenesis of Systemic Lupus Erythematosus. Rheum. Dis. Clin. 2021, 47, 427–439.
- Cooke, M.S.; Mistry, N.; Wood, C.; Herbert, K.; Lunec, J. Immunogenicity of DNA damaged by reactive oxygen species–implications for anti-DNA antibodies in lupus. Free Radic. Biol. Med. 1997, 22, 151–159.
- Hebert, S.L.; Nair, K.S. Protein and energy metabolism in type 1 diabetes. Clin. Nutr. 2010, 29, 13–17.
- Domínguez, C.; Ruiz, E.; Gussinye, M.; Carrascosa, A. Oxidative stress at onset and in early stages of type 1 diabetes in children and adolescents. Diabetes Care 1998, 21, 1736–1742.
- Gurgul-Convey, E.; Mehmeti, I.; Lortz, S.; Lenzen, S. Cytokine toxicity in insulin-producing cells is mediated by nitro-oxidative stress-induced hydroxyl radical formation in mitochondria. J. Mol. Med. 2011, 89, 785–798.
- Quiñonez-Flores, C.M.; González-Chávez, S.A.; Del Río Nájera, D.; Pacheco-Tena, C. Oxidative Stress Relevance in the Pathogenesis of the Rheumatoid Arthritis: A Systematic Review. BioMed Res. Int. 2016, 2016, 6097417.
- Tirichen, H.; Yaigoub, H.; Xu, W.; Wu, C.; Li, R.; Li, Y. Mitochondrial Reactive Oxygen Species and Their Contribution in Chronic Kidney Disease Progression Through Oxidative Stress. Front. Physiol. 2021, 12, 627837.
- Ling, X.C.; Kuo, K. Oxidative stress in chronic kidney disease. Ren. Replace. Ther. 2018, 4, 53.
- Picard, M.; McEwen, B.S.; Epel, E.S.; Sandi, C. An energetic view of stress: Focus on mitochondria. Front. Neuroendocr. 2018, 49, 72–85.
- Yue, J.L.; Yao, H. Mitochondrial dysfunction in inflammatory responses and cellular senescence: Pathogenesis and pharmacological targets for chronic lung diseases. Br. J. Pharmacol. 2016, 173, 2305–2318.
- Barnes, P.J. Oxidative stress-based therapeutics in COPD. Redox Biol. 2020, 33, 101544.
- Schneider, J.L.; Rowe, J.H.; Garcia-de-Alba, C.; Kim, C.F.; Sharpe, A.H.; Haigis, M.C. The aging lung: Physiology, disease, and 465 immunity. Cell 2021, 184, 1990–2019.
- Rahman, I.; Biswas, S.K.; Kode, A. Oxidant and antioxidant balance in the airways and airway diseases. Eur. J. Pharmacol. 2006, 533, 222–239.
- Wiegman, C.H.; Michaeloudes, C.; Haji, G.; Narang, P.; Clarke, C.J.; Russell, K.E.; Bao, W.; Pavlidis, S.; Barnes, P.J.; Kanerva, J.; et al. Oxidative stress-induced mitochondrial dysfunction drives inflammation and airway smooth muscle remodeling in patients with chronic obstructive pulmonary disease. J. Allergy Clin. Immunol. 2015, 136, 769–780.
- Haji, G.; Wiegman, C.H.; Michaeloudes, C.; Patel, M.S.; Curtis, K.; Bhavsar, P.; Polkey, M.I.; Adcock, I.M.; Chung, K.F. Mitochondrial dysfunction in airways and quadriceps muscle of patients with chronic obstructive pulmonary disease. Respir. Res. 2020, 21, 262.
- Wallace, D.C.; Chalkia, D. Mitochondrial DNA genetics and the heteroplasmy conundrum in evolution and disease. Cold Spring Harb. Perspect. Biol. 2013, 5, a021220.
- Burtenshaw, D.; Kitching, M.; Redmond, E.M.; Megson, I.L.; Cahill, P.A. Reactive oxygen species (ROS), intimal thickening, and subclinical atherosclerotic disease. Front. Cardiovasc. Med. 2019, 6, 89.
- Peng, W.; Cai, G.; Xia, Y.; Chen, J.; Wu, P.; Wang, Z.; Li, G.; Wei, D. Mitochondrial Dysfunction in Atherosclerosis. DNA Cell Biol. 2019, 38, 597–606.
- Shemiakova, T.; Ivanova, E.; Grechko, A.V.; Gerasimova, E.V.; Sobenin, I.A.; Orekhov, A.N. Mitochondrial Dysfunction and DNA Damage in the Context of Pathogenesis of Atherosclerosis. Biomedicines 2020, 18, 166.
- Zhao, H.; Liu, Y.; Li, Z.; Song, Y.; Cai, X.; Liu, Y.; Zhang, T.; Yang, L.; Li, L.; Gao, S.; et al. Identification of essential hypertension biomarkers in human urine by non-targeted metabolomics based on UPLC-Q-TOF/MS. Clin. Chim. Acta 2018, 486, 192–198.
- Pinzón-Díaz, C.E.; Calderón-Salinas, J.V.; Rosas-Flores, M.M.; Hernández, G.; López-Betancourt, A.; Quintanar-Escorza, M.A. Eryptosis and oxidative damage in hypertensive and dyslipidemic patients. Mol. Cell. Biochem. 2018, 440, 105–113.
- Kiyuna, L.A.; Albuquerque, R.P.E.; Chen, C.H.; Mochly-Rosen, D.; Ferreira, J.C.B. Targeting mitochondrial dysfunction and oxidative stress in heart failure: Challenges and opportunities. Free Radic. Biol. Med. 2018, 129, 155–168.
- Sánchez-Rodríguez, M.A.; Mendoza-Núñez, V.M. Oxidative stress indexes for diagnosis of health or disease in humans. Oxid. Med. Cell. Long. 2019, 2019, 4128152.
- van der Pol, A.; van Gilst, W.H.; Voors, A.A.; van der Meer, P. Treating oxidative stress in heart failure: Past, present and future. Eur. J. Heart Fail. 2019, 21, 425–435.
- Li, A.; Zheng, N.; Ding, X. Mitochondrial abnormalities: A hub in metabolic syndrome-related cardiac dysfunction caused by oxidative stress. Heart Fail. Rev. 2021, 1–8.
- Xiang, D.; Liu, Y.; Zhou, S.; Zhou, E.; Wang, Y. Protective Effects of Estrogen on Cardiovascular Disease Mediated by Oxidative Stress. Oxid. Med. Cell. Longev. 2021, 28, 5523516.
- Perillo, B.; Di Donato, M.; Pezone, A.; Di Zazzo, E.; Giovannelli, P.; Galasso, G.; Castoria, G.; Migliaccio, A. ROS in cancer therapy: The bright side of the moon. Exp. Mol. Med. 2020, 52, 192–203.
- Finkel, T. Signal transduction by reactive oxygen species. J. Cell Biol. 2011, 194, 7–15.
- Sato, M.; Sato, K. Maternal inheritance of mitochondrial DNA by diverse mechanisms to eliminate paternal mitochondrial DNA. Biochim. Biophys. 2013, 1833, 1979–1984.
- Chandra, D.; Singh, K.K. Genetic insights into OXPHOS defect and its role in cancer. Biochim. Biophys. Acta 2011, 1807, 620–625.
- Di Emidio, G.; Falone, S.; Artini, P.G.; Amicarelli, F.; D’Alessandro, A.M.; Tatone, C. Mitochondrial Sirtuins in Reproduction. Antioxidants 2021, 10, 1047.
- Zahra, K.; Lefter, R.; Ali, A.; Abdellah, E.C.; Trus, C.; Ciobica, A.; Timofte, D. The Involvement of the Oxidative Stress Status in Cancer Pathology: A Double View on the Role of the Antioxidants. Oxid. Med. Cell. Longev. 2021, 5, 9965916.
- Harper, M.E.; Bevilacqua, L.; Hagopian, K.; Weindruch, R.; Ramsey, J.J. Ageing, oxidative stress, and mitochondrial uncoupling. Acta Physiol. Scand. 2004, 182, 321–331.
More