Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Rossella Miele and Version 3 by Jessie Wu.
Prokineticins are a new class of chemokine-like peptides that bind their G protein-coupled receptors, Prokineticin receptor 1 (PKR1) and prokineticin receptor 2 (PKR2), and promote chemotaxis and the production of pro-inflammatory cytokines following tissue injury or infection.
- prokineticin receptors
- prokineticins
- splicing mechanisms
1. Introduction
Prokineticinin 1 (PK1 or EG-VEGF, Endocrine Gland derived-Vascular Endothelial Growth Factor) and prokineticin 2 (PK2) are small proteins that are classified as novel chemokine-like proteins based on their sequence, structure, and functions [1].
PKs have an identical AVITG sequence (alanine, valine, isoleucine, threonine, glycine) at the N-terminal amino end, which is essential for their biological activity, 10 cysteine residues (Cys) that form 5 disulfide bridges and give the molecules a very compact structure, and a tryptophan residue (Trp) at position 24, which is crucial for binding to the receptors [2][3][4][2,3,4]. Prokineticin receptors (PKRs) are seven-transmembrane G protein-coupled receptors (GPCRs), designated PKR1 and PKR2. PKRs can signal through the Gαs and Gαi proteins, modulating adenylate cyclase, as well as through the Gq proteins, promoting intracellular Ca2+ increase and protein kinase C activation, via the phospholipase C (PLC) pathway [5].
PKs and PKRs are expressed in both the central and peripheral nervous systems [6] and in peripheral organs and tissues such as ovaries, testes, intestinal tract, heart, bone marrow, and peripheral blood, and are involved in many physiological processes such as neurogenesis, circadian rhythm regulation, gastrointestinal motility, reproduction, and angiogenesis [7]. Dysregulation of prokineticin signaling pathways may be responsible for various pathological conditions such as cancer, pain, and inflammation. Indeed, after inflammatory insults, the prokineticin system is strongly upregulated in different tissues as lymphoid organs, circulating leukocytes and hematopoietic cells, synoviocytes and dendritic cells. This contributes to induce a proinflammatory phenotype that stimulates macrophage chemotaxis and cytokine release. This maintains a positive inflammatory feedback loop that forms the basis for the development of pathological conditions. Due to their central role in inflammation and neuroinflammation, prokineticins and prokineticin receptors have been identified as potential therapeutic targets in a variety of inflammatory/neuroinflammatory diseases [8]. In addition to its role in leukocyte trafficking, PKR2 is also involved in Chagas disease, as it enables the causative pathogen, the parasite Trypanosoma cruzy, to invade and infect mammalian host cells [9]. Mutations in human pkr2 or pk2 genes have been associated with genetic syndromes such as Kallmann syndrome (KS) [10] and isolated hypogonatropic hypogonadism (IHH) [11].
Given the wide range of activities in which the prokineticin system is involved, it is not surprising that numerous mechanisms have been found to regulate the activities of both prokineticins and their receptors. These mechanisms include changes in prokineticin-receptor interactions in response to a variety of environmental stimuli, modulation of protein levels in specific tissues, and changes in their molecular structures.
2. Prokineticin Receptor 1 and Prokineticin Receptor 2
Prokineticin receptor 1 (PKR1) and prokineticin receptor 2 (PKR2) are the two identified receptors for prokineticins [12][13][14][12,13,14]. They belong to family A of G protein-coupled receptors (GPCR) with two Cys residues and show similarity to the neuropeptide Y (NPY) receptor [15]. They are approximately 85% identical with sequences that appear to be highly conserved in almost all regions except the extracellular NH2 terminus, where most sequence variation is concentrated. PKRs are differently distributed in organs and tissues: PKR1 is mainly present in peripheral tissues and in the peripheral nervous system (PNS) while PKR2 is mainly abundant in the central nervous system (CNS) [7]. Upon binding to their prokineticin ligands, the receptors undergo conformational changes that lead to the activation of intracellular effectors (G-proteins or β-arrestins) and trigger signal transduction pathways that culminate in a cellular response [16][17][16,17].
2.1. Genetic and Splicing Variants
In humans, the genes encoding prokineticin receptors are located on two different chromosomes: the pkr1 gene is located on chromosome 2 (2p13.3) and the pkr2 gene is located on chromosome 20 (20p13). In mice, the pkr genes are located on chromosome 6 and 2, respectively, and consist of three exons and two introns [15]. The first exon contains a 5′-UnTranslated Region (UTR); the second exon contains part of the 5′-UTR sequence and a region encoding the first three transmembrane domains TM1, TM2, and TM3; the third exon encodes the last transmembrane domains (TM4, TM5, TM6, and TM7) and the 3′-UTR sequence. The second intron is located at the TM3 boundary within the common DRY (Asp-Arg-Tyr) sequence (Figure 1).
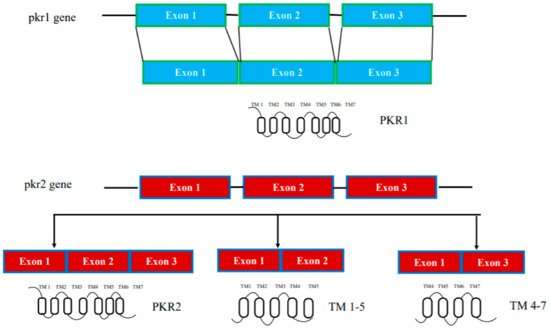
Figure 1. Schematic representation of the pkr1 and pkr2 gene structure.
The pkr2 gene is located in a region highly susceptible to mutations, as shown by linkage analysis studies and confirmed by the existence of several polymorphisms of the pkr2 gene associated with some diseases. Thus, the pkr2 polymorphism rs6053283 has been detected in idiopathic and recurrent pregnancy loss [18][19][18,19]. The presence of this rs6053283 polymorphism alters the exonic splicing enhancer (ESE) motif, which is recognized by serine/arginine-rich (SR) proteins that may be involved in the PKR2 splicing mechanism [18][19][18,19]. pkr2 gene polymorphisms have also been described in depressive and bipolar disorders and methamphetamine addiction in the Japanese population [20]. More recently, five additional polymorphisms have been found in idiopathic central precocious puberty (CPP), a syndrome characterized by premature activation of hypothalamic GnRH secretion in the absence of congenital or acquired organic lesions in the central nervous system [21].
In addition to genetically encoded mutations, the amino acid sequences of prokineticin receptors can be altered by alternative splicing mechanisms in mRNA. One PKR2 splice variant, TM 4-7, has been identified in vivo in the rat hippocampus. It consists only of the regions spanning the last three transmembrane domains (TM) and is encoded by an mRNA lacking the second exon. TM 4-7 receptor is a functional receptor because it is activated by PK2 and is able to form heterodimers with PKR2. TM 4-7 receptor isoform appears to be upregulated under pathological conditions such as in Alzheimer’s disease (AD). In the animal model of AD, induced by intracerebroventricular (i.c.v.) injection of the b amyloid peptide toxic fragment 1–42 (Aβ1–42) n the rat, this PKR2 splice variant is strongly upregulated, leading to an increase in the expression ratio between TM 4-7/PKR2 in the rat hippocampus [22][23]. The different TM 4-7 receptor levels under physiological and pathological conditions suggest a versatile way of regulating brain functions, adding another level of complexity to the prokineticin system (Figure 1).
2.2. Transcriptional Regulation
Fine transcriptional regulation allows specific expression of prokineticin receptors in different tissues and in response to a variety of stimuli. In the ovaries, PKR1 is regulated by hypoxia [23][34]. Indeed, PKR1 appears to be strongly upregulated by a hypoxic environment in the first trimester of gestation [24][35]. In particular, a further PKR1 increase is induced by human chorionic gonadotropin (βhCG) between the 8th and 11th week of gestation [25][36]. High levels of βhCG and consequently PKR1 are observed in pathological human pregnancies, such as fetal growth restriction (FGR) and pre-eclampsia (PE) [26][37]. Moreover, microarray analysis and in situ hybridization demonstrated that zinc finger homeodomain family member 1 (TSHZ1), which is essential for olfactory bulb development (OB), binds and regulates pkr2 gene expression [27][38].
2.3. Structural Elements Underlying Receptors Function
Chemokine receptors are GPCRs, integral membrane proteins consisting of seven transmembrane helical segments arranged parallel to each other and packed together in a compact bundle. The extracellular side of the receptor comprises an elongated, largely unstructured N-terminal region and three connecting loops (extracellular loops, ECL1, 2 and 3) with two conserved disulfide bonds linking the N-terminus to ECL3 and ECL1 to ECL2. The cytoplasmic side of the receptor includes three additional connecting loops (Intracellular Loops, ICL1, 2 and 3) and the C-terminal region involved in G-protein coupling [28][39].
2.3.1. Receptor-Prokineticin Interactions
Recent structures of chemokine-linked receptors confirm the two-sided model mechanism of chemokine-receptor interaction; after initial recognition of the chemokine by the N-terminus of the receptor and ECL2 (site 1), the signal is transduced by insertion of the flexible N-terminal domain of the chemokine ligand into the orthosteric pocket of the GPCR (site 2). The specific interactions with the extracellular surface of the receptor, especially with ECL2, confirm the identity of the ligand, while the insertion of the N-terminal region of the chemokine into the orthosteric TM-binding site induces conformational changes that trigger intracellular signal transduction [29][40].
The importance of ECL2 for ligand binding at site 1 of prokineticin receptors was first demonstrated by functional analysis of the PKR2 Q210R mutation identified in KS patients. The mutation has no effect on protein expression but strongly affects ligand binding capacity [30][26].
In the ECL2 of PKR2, as in all chemokine receptors, there is an aromatic residue cluster with Trp 212 localized four residues downstream of the conserved cysteine of ECL2. The crucial role of Trp at position 212 for ligand binding was demonstrated by the in vivo incorporation of p-benzoyl-L-phenylalanine, a photoactivatable unnatural amino acid, using amber codon suppression technology [31][41].
Prokineticin receptor site 2, the orthosteric TM-binding site identified by computational analysis, accepts the prokineticin amino sequence AVITG as well as unnatural small molecule ligands identified as prokineticin receptor antagonists [32][33][42,43]. Comparison between the binding sites of PKR1 and PKR2 TM revealed that they are completely conserved except for one residue: Valine (V) 207 in human PKR1, which is Phenylalanine (F) 198 in human PKR2. The F198V mutation in PKR2 allows obtaining a receptor capable of binding PK2β, a highly specific PKR1 ligand, more efficiently, demonstrating a role of this residue in ligand specificity [31][34][35][41,44,45].
2.3.2. Receptor-G-Protein Interactions
The role of intracellular loops 2 and 3 in mediating interaction with G proteins was analyzed by site-directed mutagenesis.
In PKR2, the basic amino acids of the C-terminus of ICL2, which are highly conserved in GPCRs, cooperatively bind to the C-terminal five residues of the Gαq protein. This interaction was confirmed by characterization of the PKR2 mutant R164Q identified in KS patients. Indeed, this PKR2 mutant is unable to interact with Gαq, Gαi and Gα16 proteins [36][46].
Two parts can be identified in ICL3: a proximal and a distal sequence. The proximal sequence of ICL3 is critical for G protein coupling. A mutant obtained by deletion of the arginine 264-lysine 265-arginine 266 (RKR) sequence is normally expressed on the cell surface but shows a loss of function due to a lack of G-protein coupling. In addition, the distal region of ICL3, extending from amino acids 270 to 274, is equally important for PKR2 function. The IHH-associated PKR2 R270H mutation disrupts the interaction of PKR2 with Gαq protein but not with Gαs protein.
2.4. Maturation, Folding, and Localization
GPCRs, as well as all membrane and secretory proteins, achieve proper folding and undergo post-translational modification in endoplasmtic reticulum (ER). Subsequently, the proteins enter the Golgi apparatus that lead to their final destination within the cell. However, if a polypeptide does not complete this process correctly, the incorrect protein conformation must be detected by the quality control system and remains in the ER and/or undergoes degradation [37][47].
2.4.1. Post-Translation Modifications: N-Glycosylation
The importance of N-linked glycosylation for the transport of PKRs was demonstrated, as has been done for numerous GPCRs, by creating a mutant with a replacement of asparagine at position 27 by glutamine. Unable to reach the membrane, the mutant shows a reduction in both Gαs and Gαq/11 signaling [38][48].
Mutations in ICL1 of PKR2 (R80C, R85C, and R85H) identified in patients with IHH impair N-glycosylation and localization of the receptor. PKR2 mutations R85C and R85H only slightly impair receptor function, whereas PKR2 mutation R80C leads to a marked reduction in receptor activity and exerts a dominant-negative effect on wild-type PKR2 (WT) by impairing expression of the receptor WT [39][27].
2.4.2. Dimerization
Oligomerization of GPCRs can have a major impact on receptor properties, such as ligand binding selectivity, G-protein coupling, signal transduction mechanisms, and trafficking [40][49].
PKR2 forms active dimers in physiologically relevant cell types [41][50]. By heterologous expressing PKR2 in Saccharomyces cerevisiae, reswearchers investigated the mechanisms of intermolecular interaction of PKR2 dimerization. The possible involvement of three types of mechanisms was investigated: coiled coil, disulfide bridges, and hydrophobic interactions between transmembrane domains. Characterization of several deleted or site-directed PKR2 mutants suggests that dimerization occurs through interactions between transmembrane domains, particularly TMs 4 and 5. A mutant resulting from deletion of TMs 5-7 (TM 1-4) cannot associate with WT PKR2, but it can still associate with a truncated mutant lacking TMs 6-7 (TM 1-5) via a domain swapping mechanism [41][50]. The role of the TM5 domain in modulating PKR2 functions was demonstrated by the ability of the artificial TM 1-5 mutant to exert paradoxical gain-of-function effects when co-transfected with WT PKR2 [42][24]. This result was subsequently confirmed by the identification of a heterozygous frameshift mutant of PKR2 in a 3.5-year-old girl with central precocious puberty, encoding a mutant protein corresponding to TM 1-5 (Figure 1). This mutant shows no signal transduction activity in vitro, but induces distinct ligand-induced Ca2+ responses when it heterodimerizes with WT-PKR2 [43][25].
Six PKR2 mutations (R80C, L173R, W178S, G234D, V274D, and P290S) associated with KS/IHH do not reach the cell surface and are trapped in the cellular secretory pathway. Three of these mutations (W178S, G234D, and P290S) that affect trafficking are located in the transmembrane domain and likely cause the inability to form dimeric complexes [44][32].
2.4.3. Binding to Accessory Proteins
Melanocortin Receptor Accessory Protein 2 (MRAP2) is an important regulator of energy homeostasis and its loss leads to severe obesity in rodents. MRAP2 was originally discovered to be an accessory protein of melanocortin receptor 4 (MC4R). Further experiments showed that MRAP2 and MC4R are not co-localized in different tissues and that MRAP2-KO or MC4R-KO mice do not show an overlapping phenotype [45][51]. Subsequently, other GPCRs involved in energy homeostasis have been identified as MRAP2 targets; in particular, MRAP2 significantly and specifically inhibits PKR signaling [45][46][51,52].
Specific C-terminal MRAP2 regions, conserved during evolution, are required to inhibit the activity and localization of PKRs [47][53]. In the presence of MRAP2, PKR2 glycosylation is largely prevented, blocking PKR2 to the ER [48][29].
Yeast two-hybrid screening demonstrated the interaction of snapin with the C-terminus of PKR2 (amino acids 333‣384). Snapin was originally identified as a binding protein of SNAP-25, a component of the SNARE complex, but was recently shown to also interact with the GPCR α1A-adrenoceptor [49][54]. The interactive motifs of PKR2 with snapin, YFK (343‣345) and HWR (351‣353), which share a similar sequence of two aromatic amino acids followed by a basic amino acid, have been mapped by GST pull-down and co-immunoprecipitation studies. The interaction between snapin and PKR2 did not affect PKR2 signaling but enhanced ligand-induced degradation, suggesting a role for snapin in PKR2 trafficking [50][55].
2.5. Internalization Recycling, or Degradation
In the past, PKRs have also been shown to be regulated by desensitization mechanisms. In in vitro experiments using Bv8, the amphibian homologous of PK2, Mollay [51][56] showed strong Bv8-induced tachyphylaxis during contraction of guinea pig ileum and in vivo, Cheng et al. [52][57] observed strong desensitization of PKRs after i.c.v. infusion of Bv8/PK2.
Receptor internalization is a mechanism used by most receptors to prevent diseases due to their excessive and dysregulated stimulation. The process begins with activation of the receptor by ligand and phosphorylation of the C-terminus of the receptor by G-protein receptor kinases (GRKs), resulting in desensitization. The phosphorylated receptor recruits β-arrestin, which attracts clathrin, resulting in internalization of the receptor into clathrin-coated vesicles. The receptor and ligand are recycled back to the cell membrane or transported to the lysosome for degradation [53][58].
Like many GPCRs, PKRs undergo rapid ligand-induced endocytosis followed by recycling to the plasma membrane after binding and activation by prokineticins. The mechanism underlying the internalization of PKRs is GRK2- and clathrin-mediated and independent of arrestin, as shown by Yin [17] in human embryonic kidney (HEK) cells transfected with green fluorescent protein (EGFP)-tagged PKR2 and stimulated with PK2. Indeed, in these cells, after PK2 stimulation, PKR2 recruits an unknown protein phosphorylated by GRK2 to guide the receptor to clathrin-coated pits and induces endocytosis, and on the other hand, induces Gα- and Gβγ-dissociations. The Gβγ subunit activates PLCβ and phosphorylates ERK1/2 via MEK1/2 [17].
Nevertheless, a recent paper [54][59] used the bioluminescence resonance energy transfer method (BRET) to investigate the association of PKRs with β-arrestin isoforms. This revealed that both receptors form stable BRET-emitting complexes with β-arrestin 2 but not with β-arrestin 1, indicating strong selectivity for the former. This is consistent with the presence of a β-arrestin 2 phosphosite signature at the C-tail sequence of PKR2, which has also been identified in other group A GPCRs such as 2-adrenoids and opioid receptors [54][59].