Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Camila Xu and Version 1 by Bianca Brundel.
Cytoskeletal protein variants include variants in desmin, lamin A/C, titin, myosin heavy and light chain, junctophilin, nucleoporin, nesprin, and filamin C.
- atrial fibrillation
- genetics
- cytoskeletal proteins
1. Introduction
Atrial fibrillation (AF) is the most common age-related cardiac arrhythmia in Western society [1]. AF is characterized by irregular and often very fast contractions of the atrial cardiomyocytes, resulting in an irregular heart rate, palpitations, dizziness, shortness of breath, and tiredness in the patient. AF can occur when abnormal electrical impulses suddenly start firing in the atria and override the heart’s natural pacemaker, which can no longer control the rhythm of the heart [1]. Importantly, AF is associated with severe complications, such as thromboembolic events, heart failure, cognitive impairment, and increased mortality [1]. Although in most cases, AF initially presents as short, self-terminating episodes, it often progresses into long-lasting episodes that are more difficult to reverse to sinus rhythm [1]. The progressive stages of AF are associated with structural changes that promote contractile dysfunction and the impairment of electrical conduction in the atrial myocardium [2,3,4,5,6,7][2][3][4][5][6][7]. Thus, the early detection of AF and the identification of patients at risk is of utmost importance in order to treat this arrhythmia and prevent its progression. Therefore, knowledge on the root causes of AF is essential.
Recent research investigated the potential root causes for AF. These include environmentally-induced ‘wear and tear’ AF, congenital AF, and genetic AF [1]. ‘Wear and tear’ AF is associated with the sequela of aging as well as Western diet and lifestyle-related risk factors, such as hypertension, diabetes, obesity, and coronary artery diseases, as well as various non-cardiovascular diseases, including chronic kidney disease [1]. Furthermore, an estimated prevalence of ~5% of the patients with a congenital heart disease develop AF due to a combination of flaws in embryogenesis and peri- and post-operative factors related to correction of the heart defect [8]. This so-called congenital AF is characterized by AF onset at a younger age, and these patients often rapidly progress from persistent to permanent AF [9,10][9][10]. However, not all AF patients present with predisposing ‘wear and tear’ or congenital AF. In a subset of patients who account for approximately 15% of the AF patient population, AF is familial, suggesting a genetic predisposition (Figure 1) [11,12,13,14][11][12][13][14].
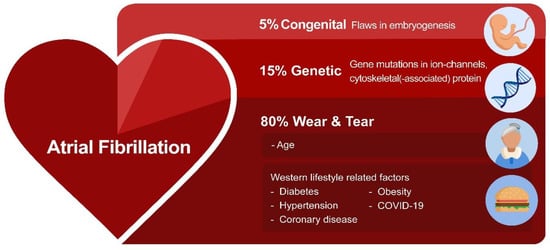
Figure 1. The variety of root causes that drive AF. In most AF patients, environmentally-induced ‘wear and tear’ related to aging or the Western lifestyle triggers AF. In addition, flaws in genetics (variants in ion channels and cytoskeletal proteins) and embryogenesis contribute to AF susceptibility.
Emerging research findings indicate a prominent role of genetic variations mainly in ion channel and cytoskeletal (-associated) genes in driving AF [15]. Although a research paper described the role of ion channel gene variants to underlie arrhythmias, including AF [16], insights into the role of cytoskeletal (-associated) protein variants as triggers for AF are in their infancy.
2. Key Role of the Cytoskeletal Network in Cardiomyocyte Function
It was only recently that the crucial importance of the cytoskeleton function to maintain balanced protein (i.e., proteostasis [3,6,17,18][3][6][17][18]) and cardiomyocyte function has been recognized. In cardiomyocytes, the cytoskeleton not only provides a communication highway by transporting proteins throughout the cell, but it also capacitates its contractile function [19,20,21][19][20][21]. In cardiomyocytes, the cytoskeleton is highly specialized, consisting of actin filaments, desmin (intermediate) filaments, and microtubules, interacting with membrane-associated proteins, sarcomeric and nuclear proteins, and proteins of the intercalated disk. This complex network, which also interacts with various organelles, including sarcoplasmic reticulum (SR), mitochondria, and the nucleus, plays an important role in the transmission of signals and the transport of (ubiquitinated) proteins within the proteostasis network [6,19,22][6][19][22] (Figure 2). Within the proteostasis network, especially the microtubules are of vital importance. As the microtubules, SR/endoplasmic reticulum (ER), sarcomeres, and mitochondria are in contact with each other, loss in contact results in Ca2+ overload in the organelles, unfolded protein response in the ER, and, consequently, excessive autophagic protein degradation and mitochondrial dysfunction [5,6,23,24,25][5][6][23][24][25]. Contact between organelles and trafficking through the cells is mediated by acetylated microtubules [3,6,26,27][3][6][26][27]. Consequently, AF-induced histone deacetylase 6 (HDAC6) activation and the subsequent deacetylation and degradation of the microtubule network have detrimental effects on the trafficking of proteins as well as on the contractile function of the atrial cardiomyocytes [3,28,29][3][28][29]. Therefore, a functional cytoskeletal network underlies a balanced communication within the proteostasis network and ensures proper contractile function. The conservation of this network is of utmost importance to ensure proper cardiac function. As such, attenuation of the disruption of the cytoskeleton protects against cardiac diseases such as AF and heart failure [3,26,30][3][26][30].
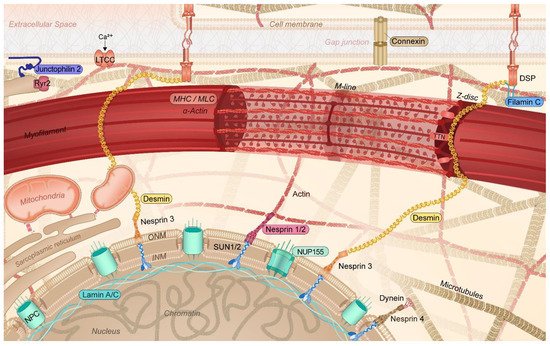
Figure 2. Schematic representation of the specific cytoskeletal protein complexes in cardiomyocytes linked to the onset of AF. Cytoskeletal (-associated) protein variants related to AF are highlighted (colored box). DSP: desmoplakin; INM: inner nuclear membrane; LTCC: L-type calcium channel; MHC: myosin heavy chain; MLC: myosin light chain; NPC: nuclear pore complex; NUP155: nucleoporin 155; Ryr2: ryanodine receptor 2; TTN: titin; ONM: outer nuclear membrane; SUN1/2: Sad1p, UNC-84 domain-containing protein 1/2.
3. Cytoskeletal (-Associated) Variants Associated with Clinical AF
In approximately 15% of AF patients, AF occurs in the absence of common risk factors and at a younger age [1]. In these patients, AF may be familial, suggesting a heritable genetic predisposition. To explore the role of gene variations in AF, the emergence of exome and genome sequencing data has provided extensive new data and revealed a previously unsuspected link between AF and several cytoskeletal (-associated) proteins (Table 1).
Recently, several AF families have been identified to carry a mutation in genes encoding the intermediate filament proteins lamin A/C (LMNA), desmin (DES), and titin (TTN) [31,32,33,34][31][32][33][34] (Table 1). Intermediate filament proteins integrate the outer cell membrane via desmoplakin (DSP), with sarcomeric proteins such as titin, Z-disk, and the nuclear membrane, thereby regulating the sarcomere architecture and function as well as the nuclear morphology, DNA stability, and gene expression (Figure 2) [6,35,36,37][6][35][36][37]. Variants in these cytoskeletal proteins are known to be associated with the development of dilated cardiomyopathy (DCM), hypertrophic cardiomyopathy (HCM), and peripartum cardiomyopathy (PPCM) [38,39,40,41][38][39][40][41]. Of note, various studies have revealed that early-onset AF often represents the initial manifestation of the cardiac phenotype, sometimes even preceding cardiomyopathy by several years, and in the absence of common risk factors as well as gross structural changes in the heart. This observation indicates that AF is a direct consequence of the mutation rather than being caused by structural abnormalities in the ventricle [42,43][42][43]. Over 50% of LMNA [31[31][42][44],42,44], 60% of DES [32] and 30–60% of TTN [34] mutant carriers develop cardiac conduction disorders, arrhythmias, and atrial tachycardia, including AF. Furthermore, these patients reveal a more progressive form of cardiac disease with poor outcomes. These findings are further supported by other studies. In a Chinese family, four subjects revealed an AV block, and three of them suffered from AF, which was related to a new frameshift insertion in the LMNA gene (c.825_826insCAGG) [45]. In addition, in a study in Italian families with LMNA variants [46], a total of 30 subjects were included, of which 19 were positive and 11 were negative for LMNA variants. Of the 19 positive study subjects, 11 showed early AF versus none of the 11 negative subjects. These studies reveal a clear association between LMNA variants and the development of AF.
Interestingly, a recent study including early-onset familial AF patients uncovered the phosphodiesterase-4D-interacting-protein (PDE4DIP) p.A123T mutation as a genetic modifier of DES p.S13F [47]. PDE4DIP p.A123T increases the penetrance of cardiac arrest and early-onset AF in the DES mutation carriers. These findings suggest an epistatic interaction of DPE4DIP with the DES gene, leading to increased penetrance of both traits and AF promotion [47].
Table 1. Overview of cytoskeletal (-associated) protein variants, identified with whole genome sequencing, that relate to genetic AF.
| Protein | Gene | Patho-Mechanism: | Main Conclusions | Refs | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Electrical | Molecular/Functional | |||||||||
| Desmin | DES | Changes AERP | Protein aggregates, PQC, autophagy. | 60% of patients exhibit conduction disease and arrhythmias, of which 9% is AF. | [32] | |||||
| Arrhythmogenic, not related to connexin redistribution. | [48] | |||||||||
| Lamin A/C | LMNA | ↓ I | Na | PQC, HSP, myolysis, nuclear blebbing, nuclear protein aggregation, altered nucleocytoplasmatic transport. | 52% of individuals with R331Q experienced AF. | [31] | ||||
| 50% of individuals with c.475G>T, p.E159* experienced AF. | [49] | |||||||||
| p.R399C associated with AF and lone AF. | [50] | |||||||||
| c.544C>T, p.Q182* associated with PAF drives progression to permanent AF. | [51] | |||||||||
| Nonsense mutation c.G1494A, p.W498* associated with AF. | [52] | |||||||||
| Titin | TTN | Abnormal ECG | Disruption sarcomeres, fibrosis (zebrafish). | Truncated titin variants associated with AF. | [53] | |||||
| Loss of function variants in titin is associated with early-onset AF. | [54] | |||||||||
| Loss of function mutation in titin is related to early-onset AF, including in ethnic minority probands. | [55] | |||||||||
| Myosin heavy chain | MYH6 | NA | Hypertrophy. | Genetic variants of MYH6 associated with AF. | [55] | |||||
| MYH7 | 47% of p.R663H mutation carriers with ventricular hypertrophy showed with AF. | [56] | ||||||||
| MYH7 | Atrial fibrosis, impairment of thick filament assembly. | Patients with p.A1379T gene mutation present extensive atrial fibrosis without clear ventricular involvement. | [57] | |||||||
| Myosin light chain | MYL4 | NA | Destabilization of F-actin—Z-disk complex. | p.E11K mutation causes early-onset AF. | [58] | |||||
| All | MYL4 | c.234delC; p.Cys78Trpfs*29 carriers showed early-onset AF. | [59] | |||||||
| Connexin 40, 43 | GJA5 | Electrical conduction changes | LOF gap junction coupling. | Association between AF and connexin 40 p.Q236H, p.K107R, p.L223M, and p.I257L variants. Connexin 40 p.Q236H mutation is linked with impaired gap junction activity. |
[60] | |||||
| Connexin 40 p.A96S mutation is associated with lower junctional conductance and enhanced sensitivity voltage gating. | [61] | |||||||||
| GJA1 | Genetic mosaicism of connexin 43 c.932delC variant in AF patient. Connexin 43 c.932delC had negative effect on gap junction formation and function. |
[62] | ||||||||
| Junctophilin 2 | JPH2 | NA | LOF impaired RyR2 stabilization, spontaneous Ca | 2+ | release. | JPH2 | p.E169K mutation is linked with higher AF inducibility via SR Ca | 2+ | by RyR2 destabilization. | [63] |
| Nucleoporin 155 | NUP155 | ECG abnormalities, APD ↓ | LOF nuclear localization, loss nuclear permeability for HSP70. | NUP155 p.R391H human mutation associated with AF. Mutant | NUP155 | mice presented reduced atrial AP duration and impaired nucleocytoplasmic transport of HSP70. | [64] | |||
| Nesprin 2 | SYNE2 | NA | SYNE is involved in RNA polymerase II binding and alternative splicing. | SYNE2 | A + 688G mutation associated with AF. | [65] | ||||
| Filamin C | FLNC | ECG abnormalities | Reduced localization at Z-disk, but preserved at intercalated disk. Diminished contractile activity. | FLNC | variants are linked with AF. | FLNC | p.V2297M mutation causes cardiomyocyte dysfunction. | [66] | ||
AERP: atrial effective refractory period; AF: atrial fibrillation; PAF: paroxysmal AF; AP: action potential; ECG: electrocardiogram LOF: loss of function; NA: not available.
Interestingly, a recent study identified multiple index patients with DCM carried an identical mutation (c.59926 + 1G>A) in the TTN gene, encoding the giant titin protein, which is the major gene underlying inherited DCM. The identified variant likely leads to truncated titan (TTNtv), which is associated with AF onset [34,53][34][53]. In an important subset (53%) of index patients and their family members carrying this founder mutation, atrial tachyarrhythmias, in particular AF, were demonstrated. In three patients, AF preceded the development of TTNtv-associated DCM by 11–14 years [34]. In patients with TTNtv-associated DCM and AF, left atrial enlargement was not noticed in half of them. In addition, the traditional risk factors for AF were absent, suggesting a potential intrinsic effect of TTNtv [34], which is in line with the study of Ahlberg et al. [53]. Together, these data strongly suggest that (paroxysmal) AF is an important part of the clinical disease spectrum caused by TTNtv, even if major structural heart abnormalities are still absent, and the individual does not present traditional risk factors for AF. However, the underlying mechanism of TTNtv causing cardiomyocyte structural and contractile remodeling and, consequently, AF remains to be elucidated.
Thus, emerging evidence has identified novel variants in cytoskeletal proteins to underlie early AF (Table 1). Currently, no detailed mechanistic explanation, treatment modalities, or diagnostic screening tools for genetic AF are available. To overcome this, we first need to understand the mutation-specific molecular pathways that drive genetic AF. This knowledge may ultimately lead to the identification of novel drug targets and therapeutic and diagnostic strategies.
References
- Hindricks, G.; Potpara, T.; Dagres, N.; Arbelo, E.; Bax, J.J.; Blomström-Lundqvist, C.; Boriani, G.; Castella, M.; Dan, G.-A.; Dilaveris, P.E.; et al. 2020 ESC Guidelines for the diagnosis and management of atrial fibrillation developed in collaboration with the European Association for Cardio-Thoracic Surgery (EACTS): The Task Force for the diagnosis and management of atrial fibrillation of the European Society of Cardiology (ESC) Developed with the special contribution of the European Heart Rhythm Association (EHRA) of the ESC. Eur. Heart J. 2021, 42, 373–498.
- De Groot, N.M.; Houden, R.P.; Smeets, J.L.; Boersma, E.; Schotten, U.; Schalij, M.J.; Crijns, H.; Allessie, M.A. Electropathological substrate of longstanding persistent atrial fibrillation in patients with structural heart disease: Epicardial breakthrough. Circulation 2010, 122, 1674–1682.
- Zhang, D.; Wu, C.T.; Qi, X.Y.; Meijering, R.A.M.; Hoogstra-Berends, F.; Tadevosyan, A.; Deniz, G.C.; Durdu, S.; Akar, A.R.; Sibon, O.C.M. Activation of Histone Deacetylase-6 (HDAC6) Induces Contractile Dysfunction through Derailment of α-Tubulin Proteostasis in Experimental and Human Atrial Fibrillation. Circulation 2014, 129, 346–358.
- Brundel, B.J.; Ausma, J.; Van Gelder, I.C.; van der Want, J.; van Gilst, W.; Crijns, H.J.; Henning, R. Activation of proteolysis by calpains and structural changes in human paroxysmal and persistent atrial fibrillation. Cardiovasc. Res. 2002, 54, 380–389.
- Wiersma, M.; Van Marion, D.M.; Wüst, R.C.; Houtkooper, R.H.; Zhang, D.; De Groot, N.M.; Henning, R.H.; Brundel, B.J. Mitochondrial Dysfunction Underlies Cardiomyocyte Remodeling in Experimental and Clinical Atrial Fibrillation. Cells 2019, 8, 1202.
- Henning, R.H.; Brundel, B. Proteostasis in cardiac health and disease. Nat. Rev. Cardiol. 2017, 14, 637–653.
- Msc, R.S.; Knops, P.; Ramos, K.S.; Roos-Serote, M.C.; Bogers, A.J.; Brundel, B.J.; Groot, N.M. Atrial fibrillation fingerprinting; spotting bio-electrical markers to early recognize atrial fibrillation by the use of a bottom-up approach (AFFIP): Rationale and design. Clin. Cardiol. 2020, 43, 546–552.
- Waldmann, V.; Laredo, M.; Abadir, S.; Mondésert, B.; Khairy, P. Atrial fibrillation in adults with congenital heart disease. Int. J. Cardiol. 2019, 287, 148–154.
- Teuwen, C.P.; De Groot, N.M. Atrial Fibrillation: The Next Epidemic for Patients With Congenital Heart Disease. J. Am. Coll. Cardiol. 2017, 70, 2949–2950.
- Teuwen, C.P.; Korevaar, T.I.; Coolen, R.L.; Van Der Wel, T.; Houck, C.A.; Evertz, R.; Yaksh, A.; Roos-Hesselink, J.W.; Bogers, A.J.; De Groot, N.M. Frequent atrial extrasystolic beats predict atrial fibrillation in patients with congenital heart defects. Europace 2016, 20, 25–32.
- Darbar, D.; Herron, K.J.; Ballew, J.D.; Jahangir, A.; Gersh, B.J.; Shen, W.-K.; Hammill, S.C.; Packer, D.L.; Olson, T.M. Familial atrial fibrillation is a genetically heterogeneous disorder. J. Am. Coll. Cardiol. 2003, 41, 2185–2192.
- Lubitz, S.A.; Yin, X.; Fontes, J.D.; Magnani, J.W.; Rienstra, M.; Pai, M.; Villalon, M.L.; Vasan, R.S.; Pencina, M.J.; Levy, D.; et al. Association Between Familial Atrial Fibrillation and Risk of New-Onset Atrial Fibrillation. JAMA 2010, 304, 2263–2269.
- Palatinus, J.A.; Das, S. Your Father and Grandfather’s Atrial Fibrillation: A Review of the Genetics of the Most Common Pathologic Cardiac Dysrhythmia. Curr. Genom. 2015, 16, 75–81.
- Tucker, N.R.; Clauss, S.; Ellinor, P.T. Common variation in atrial fibrillation: Navigating the path from genetic association to mechanism. Cardiovasc. Res. 2016, 109, 493–501.
- Zhang, J.; Johnsen, S.P.; Guo, Y.; Lip, G.Y. Epidemiology of Atrial Fibrillation: Geographic/Ecological Risk Factors, Age, Sex, Genetic. Card. Electrophysiol. Clin. 2021, 13, 1–23.
- Coppini, R.; Santini, L.; Olivotto, I.; Ackerman, M.J.; Cerbai, E. Abnormalities in sodium current and calcium homoeostasis as drivers of arrhythmogenesis in hypertrophic cardiomyopathy. Cardiovasc. Res. 2020, 116, 1585–1599.
- Wiersma, M.; Henning, R.; Brundel, B.J. Derailed Proteostasis as a Determinant of Cardiac Aging. Can. J. Cardiol. 2016, 32, 1166.e11–1166.e20.
- Meijering, R.A.; Henning, R.H.; Brundel, B.J. Reviving the protein quality control system: Therapeutic target for cardiac disease in the elderly. Trends Cardiovasc. Med. 2015, 25, 243–247.
- Knöll, R.; Buyandelger, B.; Lab, M. The Sarcomeric Z-Disc and Z-Discopathies. J. Biomed. Biotechnol. 2011, 2011, 1–12.
- Phillips, M.J.; Voeltz, G.K. Structure and function of ER membrane contact sites with other organelles. Nat. Rev. Mol. Cell Biol. 2015, 17, 69–82.
- Lewis, S.C.; Uchiyama, L.F.; Nunnari, J. ER-mitochondria contacts couple mtDNA synthesis with mitochondrial division in human cells. Science 2016, 353, aaf5549.
- Pool, L.; Wijdeveld, L.; de Groot, N.; Brundel, B. The Role of Mitochondrial Dysfunction in Atrial Fibrillation: Translation to Druggable Target and Biomarker Discovery. Int. J. Mol. Sci. 2021, 22, 8463.
- Wiersma, M.; Meijering, R.A.M.; Qi, X.; Zhang, D.; Liu, T.; Hoogstra-Berends, F.; Sibon, O.C.M.; Henning, R.H.; Nattel, S.; Brundel, B.J.J.M. Endoplasmic Reticulum Stress Is Associated With Autophagy and Cardiomyocyte Remodeling in Experimental and Human Atrial Fibrillation. J. Am. Hear. Assoc. 2017, 6, e006458.
- Ausma, J.; Dispersyn, G.D.; Duimel, H.; Thoné, F.; Donck, L.V.; A Allessie, M.; Borgers, M. Changes in Ultrastructural Calcium Distribution in Goat Atria During Atrial Fibrillation. J. Mol. Cell. Cardiol. 2000, 32, 355–364.
- Friedman, J.R.; Lackner, L.L.; West, M.; DiBenedetto, J.R.; Nunnari, J.; Voeltz, G.K. ER Tubules Mark Sites of Mitochondrial Division. Science 2011, 334, 358–362.
- Hu, X.; Li, J.; van Marion, D.M.; Zhang, D.; Brundel, B.J. Heat shock protein inducer GGA*-59 reverses contractile and structural remodeling via restoration of the microtubule network in experimental Atrial Fibrillation. J. Mol. Cell. Cardiol. 2019, 134, 86–97.
- Li, J.; Zhang, D.; Brundel, B.J.J.M.; Wiersma, M. Imbalance of ER and Mitochondria Interactions: Prelude to Cardiac Ageing and Disease? Cells 2019, 8, 1617.
- Lemon, D.D.; Horn, T.R.; Cavasin, M.A.; Jeong, M.Y.; Haubold, K.W.; Long, C.; Irwin, D.C.; McCune, S.A.; Chung, E.; Leinwand, L.; et al. Cardiac HDAC6 catalytic activity is induced in response to chronic hypertension. J. Mol. Cell. Cardiol. 2011, 51, 41–50.
- Friedman, J.R.; Webster, B.M.; Mastronarde, D.N.; Verhey, K.J.; Voeltz, G.K. ER sliding dynamics and ER-mitochondrial contacts occur on acetylated microtubules. J. Cell Biol. 2010, 190, 363–375.
- Lehmann, L.; Worst, B.C.; Stanmore, D.A.; Backs, J. Histone deacetylase signaling in cardioprotection. Cell. Mol. Life Sci. 2013, 71, 1673–1690.
- Hoorntje, E.T.; Bollen, I.A.; Barge-Schaapveld, D.Q.; Van Tienen, F.H.; Meerman, G.J.T.; Jansweijer, J.A.; Van Essen, A.J.; Volders, P.G.; Constantinescu, A.A.; Akker, P.C.V.D.; et al. Lamin A/C-Related Cardiac Disease: Late Onset With a Variable and Mild Phenotype in a Large Cohort of Patients With the Lamin A/C p.(Arg331Gln) Founder Mutation. Circ. Cardiovasc. Genet. 2017, 10, e001631.
- Van Spaendonck-Zwarts, K.Y.; van Hessem, L.; Jongbloed, J.D.; de Walle, H.; Capetanaki, Y.; van der Kooi, A.; van Langen, I.; Berg, M.V.D.; van Tintelen, J. Desmin-related myopathy. Clin. Genet. 2010, 80, 354–366.
- Karmouch, J.; Zhou, Q.Q.; Miyake, C.Y.; Lombardi, R.; Kretzschmar, K.; Bannier-Hélaouët, M.; Clevers, H.; Wehrens, X.; Willerson, J.T.; Marian, A.J. Distinct Cellular Basis for Early Cardiac Arrhythmias, the Cardinal Manifestation of Arrhythmogenic Cardiomyopathy, and the Skin Phenotype of Cardiocutaneous Syndromes. Circ. Res. 2017, 121, 1346–1359.
- Hoorntje, E.T.; Van Spaendonck-Zwarts, K.Y.; Rijdt, W.P.T.; Boven, L.; Vink, A.; Van Der Smagt, J.J.; Asselbergs, F.; Van Wijngaarden, J.; Hennekam, E.A.; Pinto, Y.M.; et al. The first titin (c.59926 + 1G > A) founder mutation associated with dilated cardiomyopathy. Eur. J. Hear. Fail. 2017, 20, 803–806.
- Ho, C.Y.; Jaalouk, D.E.; Vartiainen, M.K.; Lammerding, J. Lamin A/C and emerin regulate MKL1–SRF activity by modulating actin dynamics. Nature 2013, 497, 507–511.
- Macquart, C.; Jüttner, R.; Rodriguez, B.M.; Le Dour, C.; Lefebvre, F.; Chatzifrangkeskou, M.; Schmitt, A.; Gotthardt, M.; Bonne, G.; Muchir, A. Microtubule cytoskeleton regulates Connexin 43 localization and cardiac conduction in cardiomyopathy caused by mutation in A-type lamins gene. Hum. Mol. Genet. 2018, 28, 4043–4052.
- Barateau, A.; Vadrot, N.; Vicart, P.; Ferreiro, A.; Mayer, M.; Héron, D.; Vigouroux, C.; Buendia, B. A Novel Lamin A Mutant Responsible for Congenital Muscular Dystrophy Causes Distinct Abnormalities of the Cell Nucleus. PLoS ONE 2017, 12, e0169189.
- Tsikitis, M.; Galata, Z.; Mavroidis, M.; Psarras, S.; Capetanaki, Y. Intermediate filaments in cardiomyopathy. Biophys. Rev. 2018, 10, 1007–1031.
- Vignier, N.; Chatzifrangkeskou, M.; Rodriguez, B.M.; Mericskay, M.; Mougenot, N.; Wahbi, K.; Bonne, G.; Muchir, A. Rescue of biosynthesis of nicotinamide adenine dinucleotide protects the heart in cardiomyopathy caused by lamin A/C gene mutation. Hum. Mol. Genet. 2018, 27, 3870–3880.
- Stergiopoulos, K.; Lima, F.V.; Yang, J.; Biteker, M.; Ware, J.S.; Seidman, J.G.; Arany, Z. Shared Genetic Predisposition in Peripartum and Dilated Cardiomyopathies. N. Engl. J. Med. 2016, 374, 2601–2602.
- Pfeffer, T.J.; Schlothauer, S.; Pietzsch, S.; Schaufelberger, M.; Auber, B.; Ricke-Hoch, M.; List, M.; Berliner, D.; Moulig, V.A.; König, T.; et al. Increased Cancer Prevalence in Peripartum Cardiomyopathy. JACC CardioOncol. 2019, 1, 196–205.
- Van Tintelen, J.P.; Hofstra, R.M.W.; Katerberg, H.; Rossenbacker, T.; Wiesfeld, A.C.P.; du Marchie Sarvaas, G.J.; Wilde, A.A.M.; van Langen, I.M.; Nannenberg, E.A.; van der Kooi, A.J.; et al. High yield of LMNA mutations in patients with dilated cardiomyopathy and/or conduction disease referred to cardiogenetics outpatient clinics. Am. Heart J. 2007, 154, 1130–1139.
- Kumar, S.; Baldinger, S.H.; Gandjbakhch, E.; Maury, P.; Sellal, J.-M.; Androulakis, A.F.; Waintraub, X.; Charron, P.; Rollin, A.; Richard, P.; et al. Long-Term Arrhythmic and Nonarrhythmic Outcomes of Lamin A/C Mutation Carriers. J. Am. Coll. Cardiol. 2016, 68, 2299–2307.
- Hasselberg, N.E.; Haland, T.F.; Saberniak, J.; Brekke, P.; Berge, K.E.; Leren, T.P.; Edvardsen, T.; Haugaa, K. Lamin A/C cardiomyopathy: Young onset, high penetrance, and frequent need for heart transplantation. Eur. Hear. J. 2017, 39, 853–860.
- Jia, Z.; Zhang, Y.; Deng, J.; Guo, Y.; Du, Y.; Wang, G.; Xu, J.; Li, X. A novel LMNA indel mutation identified in a family with atrioventricular block and atrial fibrillation. Medicine 2021, 100, e25910.
- Gerbino, A.; Forleo, C.; Milano, S.; Piccapane, F.; Procino, G.; Pepe, M.; Piccolo, M.; Guida, P.; Resta, N.; Favale, S.; et al. Pro-inflammatory cytokines as emerging molecular determinants in cardiolaminopathies. J. Cell. Mol. Med. 2021.
- Ziki, M.D.A.; Bhat, N.; Neogi, A.; Driscoll, T.P.; Ugwu, N.; Liu, Y.; Smith, E.; Abboud, J.M.; Chouairi, S.; Schwartz, M.A.; et al. Epistatic interaction of PDE4DIP and DES mutations in familial atrial fibrillation with slow conduction. Hum. Mutat. 2021, 42, 1279–1293.
- Schrickel, J.W.; Stöckigt, F.; Krzyzak, W.; Paulin, D.; Li, Z.; Lübkemeier, I.; Fleischmann, B.; Sasse, P.; Linhart, M.; Lewalter, T.; et al. Cardiac conduction disturbances and differential effects on atrial and ventricular electrophysiological properties in desmin deficient mice. J. Interv. Card. Electrophysiol. 2010, 28, 71–80.
- Yokokawa, T.; Ichimura, S.; Hijioka, N.; Kaneshiro, T.; Yoshihisa, A.; Kunii, H.; Nakazato, K.; Ishida, T.; Suzuki, O.; Ohno, S.; et al. Case reports of a c.475G>T, p.E159* lamin A/C mutation with a family history of conduction disorder, dilated cardiomyopathy and sudden cardiac death. BMC Cardiovasc. Disord. 2019, 19, 298.
- Han, M.; Zhao, M.; Cheng, C.; Huang, Y.; Han, S.; Li, W.; Tu, X.; Luo, X.; Yu, X.; Liu, Y.; et al. Lamin A mutation impairs interaction with nucleoporin NUP155 and disrupts nucleocytoplasmic transport in atrial fibrillation. Hum. Mutat. 2018, 40, 310–325.
- Glöcklhofer, C.R.; Steinfurt, J.; Franke, G.; Hoppmann, A.; Glantschnig, T.; Perez-Feliz, S.; Alter, S.; Fischer, J.; Brunner, M.; Rainer, P.P.; et al. A novel LMNA nonsense mutation causes two distinct phenotypes of cardiomyopathy with high risk of sudden cardiac death in a large five-generation family. Europace 2018, 20, 2003–2013.
- Zhao, J.; Yao, H.; Li, Z.; Wang, L.; Liu, G.; Wang, D.W.; Liang, Z. A novel nonsense mutation in LMNA gene identified by Exome Sequencing in an atrial fibrillation family. Eur. J. Med. Genet. 2016, 59, 396–400.
- Ahlberg, G.; Refsgaard, L.; Lundegaard, P.R.; Andreasen, L.; Ranthe, M.F.; Linscheid, N.; Nielsen, J.B.; Melbye, M.; Haunsø, S.; Sajadieh, A.; et al. Rare truncating variants in the sarcomeric protein titin associate with familial and early-onset atrial fibrillation. Nat. Commun. 2018, 9, 4316.
- Choi, S.H.; Weng, L.-C.; Roselli, C.; Lin, H.; Haggerty, C.; Shoemaker, M.B.; Barnard, J.; Arking, D.E.; Chasman, D.I.; Albert, C.; et al. Association Between Titin Loss-of-Function Variants and Early-Onset Atrial Fibrillation. JAMA 2018, 320, 2354–2364.
- Chalazan, B.; Mol, D.; Darbar, F.A.; Ornelas-Loredo, A.; Al-Azzam, B.; Chen, Y.; Tofovic, D.; Sridhar, A.; Alzahrani, Z.; Ellinor, P.; et al. Association of Rare Genetic Variants and Early-Onset Atrial Fibrillation in Ethnic Minority Individuals. JAMA Cardiol. 2021, 6, 811.
- Gruver, E.; Fatkin, D.; Dodds, G.; Kisslo, J.; Maron, B.J.; Seidman, J.; E Seidman, C. Familial hypertrophic cardiomyopathy and atrial fibrillation caused by Arg663His beta–cardiac myosin heavy chain mutation. Am. J. Cardiol. 1999, 83, 13–18.
- Zhang, S.; Wilson, J.; Madani, M.; Feld, G.; Greenberg, B. Atrial Arrhythmias and Extensive Left Atrial Fibrosis as the Initial Presentation of MYH7 Gene Mutation. JACC Clin. Electrophysiol. 2018, 4, 1488–1490.
- Orr, N.; Arnaout, R.; Gula, L.J.; Spears, D.; Leong-Sit, P.; Li, Q.; Tarhuni, W.; Reischauer, S.; Chauhan, V.S.; Borkovich, M.; et al. A mutation in the atrial-specific myosin light chain gene (MYL4) causes familial atrial fibrillation. Nat. Commun. 2016, 7, 11303.
- Gudbjartsson, D.F.; Helgason, H.; Gudjonsson, S.A.; Zink, F.; Oddsson, A.; Gylfason, A.; Besenbacher, S.; Magnusson, G.; Halldórsson, B.; Hjartarson, E.; et al. Large-scale whole-genome sequencing of the Icelandic population. Nat. Genet. 2015, 47, 435–444.
- Noureldin, M.; Chen, H.; Bai, D. Functional Characterization of Novel Atrial Fibrillation-Linked GJA5 (Cx40) Mutants. Int. J. Mol. Sci. 2018, 19, 977.
- Lübkemeier, I.; Andrié, R.; Lickfett, L.; Bosen, F.; Stöckigt, F.; Dobrowolski, R.; Draffehn, A.M.; Fregeac, J.; Schultze, J.L.; Bukauskas, F.F.; et al. The Connexin40A96S mutation from a patient with atrial fibrillation causes decreased atrial conduction velocities and sustained episodes of induced atrial fibrillation in mice. J. Mol. Cell. Cardiol. 2013, 65, 19–32.
- Thibodeau, I.L.; Xu, J.; Li, Q.; Liu, G.; Lam, K.; Veinot, J.P.; Birnie, D.H.; Jones, D.L.; Krahn, A.D.; Lemery, R.; et al. Paradigm of Genetic Mosaicism and Lone Atrial Fibrillation. Circulation 2010, 122, 236–244.
- Beavers, D.L.; Wang, W.; Ather, S.; Voigt, N.; Garbino, A.; Dixit, S.S.; Landstrom, A.; Li, N.; Wang, Q.; Olivotto, I.; et al. Mutation E169K in Junctophilin-2 Causes Atrial Fibrillation Due to Impaired RyR2 Stabilization. J. Am. Coll. Cardiol. 2013, 62, 2010–2019.
- Zhang, X.; Chen, S.; Yoo, S.; Chakrabarti, S.; Zhang, T.; Ke, T.; Oberti, C.; Yong, S.L.; Fang, F.; Li, L.; et al. Mutation in Nuclear Pore Component NUP155 Leads to Atrial Fibrillation and Early Sudden Cardiac Death. Cell 2008, 135, 1017–1027.
- Tsai, C.-T.; Hsieh, C.-S.; Chang, S.-N.; Chuang, E.Y.; Juang, J.-M.J.; Lin, L.-Y.; Lai, L.-P.; Hwang, J.-J.; Chiang, F.-T.; Lin, J.-L. Next-generation sequencing of nine atrial fibrillation candidate genes identified novel de novo mutations in patients with extreme trait of atrial fibrillation. J. Med. Genet. 2014, 52, 28–36.
- Tucker, N.R.; McLellan, M.A.; Hu, D.; Ye, J.; Parsons, V.A.; Mills, R.W.; Clauss, S.; Dolmatova, E.; Shea, M.A.; Milan, D.J.; et al. Novel Mutation in FLNC (Filamin C) Causes Familial Restrictive Cardiomyopathy. Circ. Cardiovasc. Genet. 2017, 10.
More