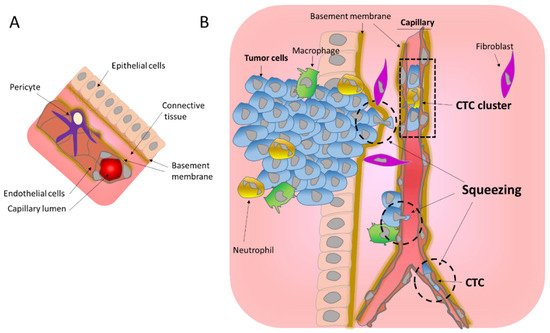
Metastasis is the process that allows the seeding of tumor cells in a new organ. The migration and invasion of cancer cells involves the pulling, pushing, and squeezing of cells through narrow spaces and pores. Tumor cells need to cross several physical barriers, such as layers of basement membranes as well as the endothelium wall during the way in and out of the blood stream, to reach the new organ.
- metastasis
- nuclear envelope rupture (NER)
- cGAS/STING
- mechanosensitivity
1. Introduction
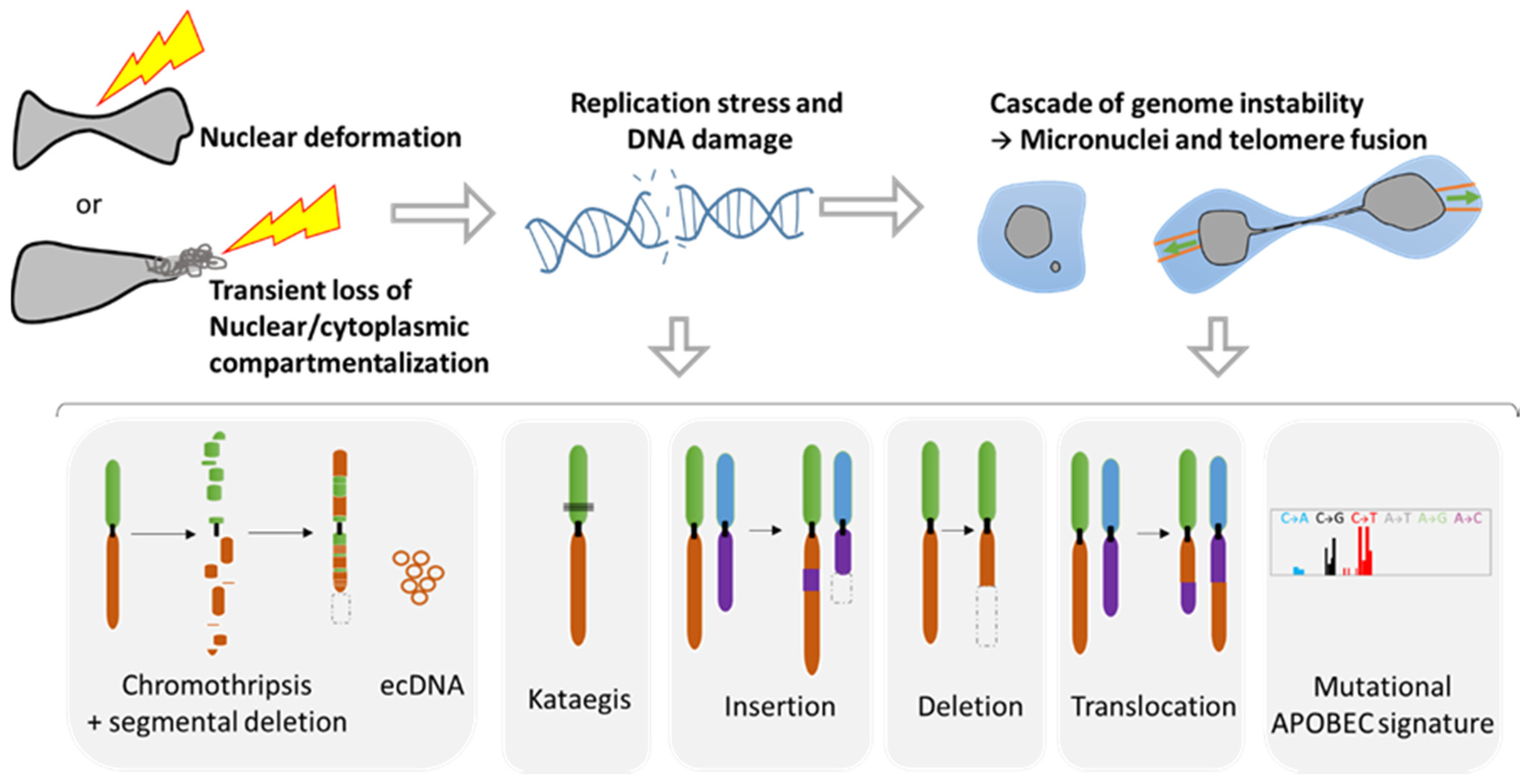
During metastasis, invading tumor cells and circulating tumor cells (CTC) face multiple mechanical challenges during migration through narrow pores and cell squeezing. However, little is known on the importance and consequences of mechanical stress for tumor progression and success in invading a new organ. Recently, several studies have shown that cell constriction can lead to nuclear envelope rupture (NER) during interphase. This loss of proper nuclear compartmentalization has a profound effect on the genome, being a key driver for the genome evolution needed for tumor progression. More than just being a source of genomic alterations, the transient nuclear envelope collapse can also support metastatic growth by several mechanisms involving the innate immune response cGAS/STING pathway.
2. Migration through the Basement Membrane

3. Consequences of Nuclear Envelope Rupture on Tumor Genomic Heterogeneity
3.1. Role of NER in Simple and Complex Chromosomal Rearrangements

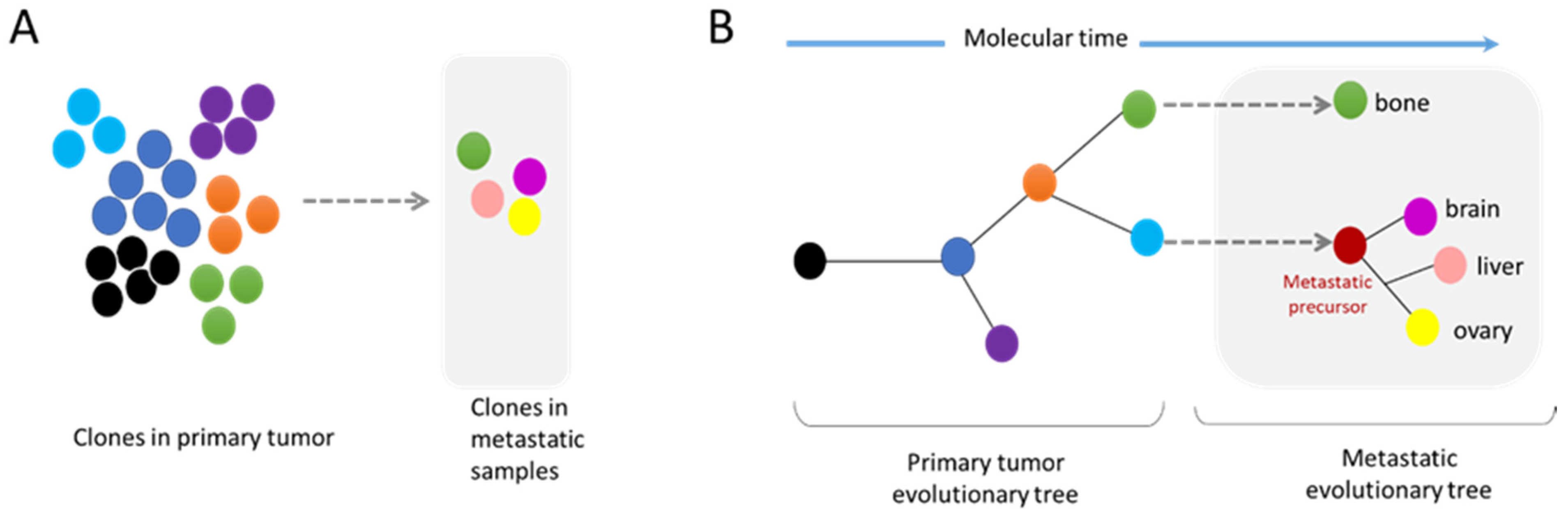
3.2. Metastasis and Genomic Evolution
4. Nuclear Squeezing and Its Role in Activating the Innate Immune Response cGAS
References
- Reuten, R.; Zendehroud, S.; Nicolau, M.; Fleischhauer, L.; Laitala, A.; Kiderlen, S.; Nikodemus, D.; Wullkopf, L.; Nielsen, S.R.; McNeilly, S.; et al. Basement membrane stiffness determines metastases formation. Nat. Mater. 2021, 20, 892–903.
- Yurchenco, P.D. Basement membranes: Cell scaffoldings and signaling platforms. Cold Spring Harb. Perspect. Biol. 2011, 3, a004911.
- Linder, S. Invadosomes at a glance. J. Cell Sci. 2009, 122, 3009–3013.
- Clark, E.S.; Weaver, A.M. A new role for cortactin in invadopodia: Regulation of protease secretion. Eur. J. Cell Biol. 2008, 87, 581–590.
- Mondal, C.; Di Martino, J.S.; Bravo-Cordero, J.J. Actin dynamics during tumor cell dissemination. Int. Rev. Cell Mol. Biol. 2021, 360, 65–98.
- Sabeh, F.; Shimizu-Hirota, R.; Weiss, S.J. Protease-dependent versus -independent cancer cell invasion programs: Three-dimensional amoeboid movement revisited. J. Cell. Biol. 2009, 185, 11–19.
- Sznurkowska, M.K.; Aceto, N. The gate to metastasis: Key players in cancer cell intravasation. FEBS J. 2021.
- Wyckoff, J.B.; Jones, J.G.; Condeelis, J.S.; Segall, J.E. A critical step in metastasis: In vivo analysis of intravasation at the primary tumor. Cancer Res. 2000, 60, 2504–2511.
- Reymond, N.; d’Agua, B.B.; Ridley, A.J. Crossing the endothelial barrier during metastasis. Nat. Rev. Cancer 2013, 13, 858–870.
- Wisdom, K.M.; Adebowale, K.; Chang, J.; Lee, J.Y.; Nam, S.; Desai, R.; Rossen, N.S.; Rafat, M.; West, R.B.; Hodgson, L.; et al. Matrix mechanical plasticity regulates cancer cell migration through confining microenvironments. Nat. Commun. 2018, 9, 4144.
- Hurtado, P.; Martinez-Pena, I.; Pineiro, R. Dangerous Liaisons: Circulating Tumor Cells (CTCs) and Cancer-Associated Fibroblasts (CAFs). Cancers 2020, 12, 2861.
- Glentis, A.; Oertle, P.; Mariani, P.; Chikina, A.; El Marjou, F.; Attieh, Y.; Zaccarini, F.; Lae, M.; Loew, D.; Dingli, F.; et al. Cancer-associated fibroblasts induce metalloprotease-independent cancer cell invasion of the basement membrane. Nat. Commun. 2017, 8, 924.
- Chang, J.; Chaudhuri, O. Beyond proteases: Basement membrane mechanics and cancer invasion. J. Cell. Biol. 2019, 218, 2456–2469.
- Denais, C.M.; Gilbert, R.M.; Isermann, P.; McGregor, A.L.; te Lindert, M.; Weigelin, B.; Davidson, P.M.; Friedl, P.; Wolf, K.; Lammerding, J. Nuclear envelope rupture and repair during cancer cell migration. Science 2016, 352, 353–358.
- Raab, M.; Gentili, M.; de Belly, H.; Thiam, H.R.; Vargas, P.; Jimenez, A.J.; Lautenschlaeger, F.; Voituriez, R.; Lennon-Dumenil, A.M.; Manel, N.; et al. ESCRT III repairs nuclear envelope ruptures during cell migration to limit DNA damage and cell death. Science 2016, 352, 359–362.
- Nader, G.P.F.; Aguera-Gonzalez, S.; Routet, F.; Gratia, M.; Maurin, M.; Cancila, V.; Cadart, C.; Palamidessi, A.; Ramos, R.N.; San Roman, M.; et al. Compromised nuclear envelope integrity drives TREX1-dependent DNA damage and tumor cell invasion. Cell 2021, 184, 5230–5246.
- Shah, P.; Hobson, C.M.; Cheng, S.; Colville, M.J.; Paszek, M.J.; Superfine, R.; Lammerding, J. Nuclear Deformation Causes DNA Damage by Increasing Replication Stress. Curr. Biol. 2021, 31, 753–765.
- Pfeifer, C.R.; Xia, Y.; Zhu, K.; Liu, D.; Irianto, J.; Garcia, V.M.M.; Millan, L.M.S.; Niese, B.; Harding, S.; Deviri, D.; et al. Constricted migration increases DNA damage and independently represses cell cycle. Mol. Biol. Cell 2018, 29, 1948–1962.
- Gauthier, B.R.; Comaills, V. Nuclear Envelope Integrity in Health and Disease: Consequences on Genome Instability and Inflammation. Int. J. Mol. Sci. 2021, 22, 7281.
- Voronina, N.; Wong, J.K.L.; Hubschmann, D.; Hlevnjak, M.; Uhrig, S.; Heilig, C.E.; Horak, P.; Kreutzfeldt, S.; Mock, A.; Stenzinger, A.; et al. The landscape of chromothripsis across adult cancer types. Nat. Commun. 2020, 11, 2320.
- Shoshani, O.; Brunner, S.F.; Yaeger, R.; Ly, P.; Nechemia-Arbely, Y.; Kim, D.H.; Fang, R.; Castillon, G.A.; Yu, M.; Li, J.S.Z.; et al. Chromothripsis drives the evolution of gene amplification in cancer. Nature 2021, 591, 137–141.
- Ly, P.; Brunner, S.F.; Shoshani, O.; Kim, D.H.; Lan, W.; Pyntikova, T.; Flanagan, A.M.; Behjati, S.; Page, D.C.; Campbell, P.J.; et al. Chromosome segregation errors generate a diverse spectrum of simple and complex genomic rearrangements. Nat. Genet. 2019, 51, 705–715.
- Crasta, K.; Ganem, N.J.; Dagher, R.; Lantermann, A.B.; Ivanova, E.V.; Pan, Y.; Nezi, L.; Protopopov, A.; Chowdhury, D.; Pellman, D. DNA breaks and chromosome pulverization from errors in mitosis. Nature 2012, 482, 53–58.
- Zhang, C.Z.; Spektor, A.; Cornils, H.; Francis, J.M.; Jackson, E.K.; Liu, S.; Meyerson, M.; Pellman, D. Chromothripsis from DNA damage in micronuclei. Nature 2015, 522, 179–184.
- Umbreit, N.T.; Zhang, C.Z.; Lynch, L.D.; Blaine, L.J.; Cheng, A.M.; Tourdot, R.; Sun, L.; Almubarak, H.F.; Judge, K.; Mitchell, T.J.; et al. Mechanisms generating cancer genome complexity from a single cell division error. Science 2020, 368, eaba0712.
- Maciejowski, J.; Hatch, E.M. Nuclear Membrane Rupture and Its Consequences. Annu. Rev. Cell Dev. Biol. 2020, 36, 85–114.
- Maciejowski, J.; Li, Y.; Bosco, N.; Campbell, P.J.; de Lange, T. Chromothripsis and Kataegis Induced by Telomere Crisis. Cell 2015, 163, 1641–1654.
- Maciejowski, J.; Chatzipli, A.; Dananberg, A.; Chu, K.; Toufektchan, E.; Klimczak, L.J.; Gordenin, D.A.; Campbell, P.J.; de Lange, T. APOBEC3-dependent kataegis and TREX1-driven chromothripsis during telomere crisis. Nat. Genet. 2020, 52, 884–890.
- Vietri, M.; Schultz, S.W.; Bellanger, A.; Jones, C.M.; Petersen, L.I.; Raiborg, C.; Skarpen, E.; Pedurupillay, C.R.J.; Kjos, I.; Kip, E.; et al. Unrestrained ESCRT-III drives micronuclear catastrophe and chromosome fragmentation. Nat. Cell Biol. 2020, 22, 856–867.
- Hatch, E.M.; Fischer, A.H.; Deerinck, T.J.; Hetzer, M.W. Catastrophic nuclear envelope collapse in cancer cell micronuclei. Cell 2013, 154, 47–60.
- Zou, J.; Wang, C.; Ma, X.; Wang, E.; Peng, G. APOBEC3B, a molecular driver of mutagenesis in human cancers. Cell Biosci. 2017, 7, 29.
- Comaills, V.; Kabeche, L.; Morris, R.; Buisson, R.; Yu, M.; Madden, M.W.; LiCausi, J.A.; Boukhali, M.; Tajima, K.; Pan, S.; et al. Genomic Instability Is Induced by Persistent Proliferation of Cells Undergoing Epithelial-to-Mesenchymal Transition. Cell Rep. 2016, 17, 2632–2647.
- Leibowitz, M.L.; Papathanasiou, S.; Doerfler, P.A.; Blaine, L.J.; Sun, L.; Yao, Y.; Zhang, C.Z.; Weiss, M.J.; Pellman, D. Chromothripsis as an on-target consequence of CRISPR-Cas9 genome editing. Nat. Genet. 2021, 53, 895–905.
- Maura, F.; Bolli, N.; Angelopoulos, N.; Dawson, K.J.; Leongamornlert, D.; Martincorena, I.; Mitchell, T.J.; Fullam, A.; Gonzalez, S.; Szalat, R.; et al. Genomic landscape and chronological reconstruction of driver events in multiple myeloma. Nat. Commun. 2019, 10, 3835.
- Bakhoum, S.F.; Ngo, B.; Laughney, A.M.; Cavallo, J.A.; Murphy, C.J.; Ly, P.; Shah, P.; Sriram, R.K.; Watkins, T.B.K.; Taunk, N.K.; et al. Chromosomal instability drives metastasis through a cytosolic DNA response. Nature 2018, 553, 467–472.
- Watkins, T.B.K.; Lim, E.L.; Petkovic, M.; Elizalde, S.; Birkbak, N.J.; Wilson, G.A.; Moore, D.A.; Gronroos, E.; Rowan, A.; Dewhurst, S.M.; et al. Pervasive chromosomal instability and karyotype order in tumour evolution. Nature 2020, 587, 126–132.
- Brown, D.; Smeets, D.; Szekely, B.; Larsimont, D.; Szasz, A.M.; Adnet, P.Y.; Rothe, F.; Rouas, G.; Nagy, Z.I.; Farago, Z.; et al. Phylogenetic analysis of metastatic progression in breast cancer using somatic mutations and copy number aberrations. Nat. Commun. 2017, 8, 14944.
- Birkbak, N.J.; McGranahan, N. Cancer Genome Evolutionary Trajectories in Metastasis. Cancer Cell 2020, 37, 8–19.
- Priestley, P.; Baber, J.; Lolkema, M.P.; Steeghs, N.; de Bruijn, E.; Shale, C.; Duyvesteyn, K.; Haidari, S.; van Hoeck, A.; Onstenk, W.; et al. Pan-cancer whole-genome analyses of metastatic solid tumours. Nature 2019, 575, 210–216.
- Klotz, R.; Thomas, A.; Teng, T.; Han, S.M.; Iriondo, O.; Li, L.; Restrepo-Vassalli, S.; Wang, A.; Izadian, N.; MacKay, M.; et al. Circulating Tumor Cells Exhibit Metastatic Tropism and Reveal Brain Metastasis Drivers. Cancer Discov. 2020, 10, 86–103.
- Ablasser, A.; Chen, Z.J. cGAS in action: Expanding roles in immunity and inflammation. Science 2019, 363, eaat8657.
- Ragu, S.; Matos-Rodrigues, G.; Lopez, B.S. Replication Stress, DNA Damage, Inflammatory Cytokines and Innate Immune Response. Genes 2020, 11, 409.
- Schadt, L.; Sparano, C.; Schweiger, N.A.; Silina, K.; Cecconi, V.; Lucchiari, G.; Yagita, H.; Guggisberg, E.; Saba, S.; Nascakova, Z.; et al. Cancer-Cell-Intrinsic cGAS Expression Mediates Tumor Immunogenicity. Cell Rep. 2019, 29, 1236–1248.
- Wang, H.; Hu, S.; Chen, X.; Shi, H.; Chen, C.; Sun, L.; Chen, Z.J. cGAS is essential for the antitumor effect of immune checkpoint blockade. Proc. Natl. Acad. Sci. USA 2017, 114, 1637–1642.
- Nassour, J.; Radford, R.; Correia, A.; Fuste, J.M.; Schoell, B.; Jauch, A.; Shaw, R.J.; Karlseder, J. Autophagic cell death restricts chromosomal instability during replicative crisis. Nature 2019, 565, 659–663.
- Gonzalez-Meljem, J.M.; Apps, J.R.; Fraser, H.C.; Martinez-Barbera, J.P. Paracrine roles of cellular senescence in promoting tumourigenesis. Br. J. Cancer 2018, 118, 1283–1288.
- Kwon, J.; Bakhoum, S.F. The Cytosolic DNA-Sensing cGAS-STING Pathway in Cancer. Cancer Discov. 2020, 10, 26–39.
- Li, J.; Duran, M.A.; Dhanota, N.; Chatila, W.K.; Bettigole, S.E.; Kwon, J.; Sriram, R.K.; Humphries, M.P.; Salto-Tellez, M.; James, J.A.; et al. Metastasis and Immune Evasion from Extracellular cGAMP Hydrolysis. Cancer Discov. 2021, 11, 1212–1227.
- Chen, Q.; Boire, A.; Jin, X.; Valiente, M.; Er, E.E.; Lopez-Soto, A.; Jacob, L.; Patwa, R.; Shah, H.; Xu, K.; et al. Carcinoma-astrocyte gap junctions promote brain metastasis by cGAMP transfer. Nature 2016, 533, 493–498.
- Yang, H.; Wang, H.; Ren, J.; Chen, Q.; Chen, Z.J. cGAS is essential for cellular senescence. Proc. Natl. Acad. Sci. USA 2017, 114, E4612–E4620.
- Dou, Z.; Ghosh, K.; Vizioli, M.G.; Zhu, J.; Sen, P.; Wangensteen, K.J.; Simithy, J.; Lan, Y.; Lin, Y.; Zhou, Z.; et al. Cytoplasmic chromatin triggers inflammation in senescence and cancer. Nature 2017, 550, 402–406.