Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 4 by Vivi Li and Version 3 by Vivi Li.
Under pressure of 1–100 GPa, unsaturated organic molecules tend to form covalent bond to each other for a negative enthalpy change, which often produces polymeric materials with extended carbon skeleton. The polymerization reactions typically happen in crystal, which promotes the topochemical process.
- high pressure
- polymerization
- topochemical reaction
- reaction mechanism
- hydrogen transfer
1. Introduction
Chemical reactions under high pressure (HP) are often related to high temperature (HT), since, under high pressure, the migration of atoms is hindered, and extra energy is required to overcome the energy barrier to reach the thermodynamic stable state, that is, the minimum point on the potential surface. This is what proceeds in most high-pressure experiments currently. However, much more meta-stable structures are remaining at the local minimums on the potential surface, waiting to be explored. The most famous examples are diamond and its analogues [1][2][3]. The HP-HT synthesis can easily synthesize cubic diamond as it is the thermodynamic stable phase at the P-T condition, but not for its metastable analogues with other sp3-C-C connection topologies.
This is similar to the traditional HT solid state reactions and molecular/cluster solution reactions at ambient pressure (AP). The dedicatedly designed solution reactions can drive the small molecules to travel on the potential surface, control the directions, and build up highly complex structures (bottom-up) with complicated physical and chemical functions. To do the same thing under HP and precisely synthesize the required metastable materials, a bottom-up process is also needed, including choosing suitable building blocks such as molecules and/or clusters, and the techniques to connect the building blocks.
For HP reaction, as mentioned above, the migration of molecules is hindered, and HP becomes the most important driving force to the reaction. This brings two characteristics. First, thermodynamically, the ΔPV term affects the enthalpy change and free energy change of the reaction significantly, and the reaction with a negative volume change is more preferred. In practice, molecules always tend to polymerize upon compression, which is referred as pressure-induced polymerization (PIP). Researchers have provided a schematic quantitative thermodynamic explanation and discussions on this in a very recent paper and in previous reviews [4][5][6][7][8][9]. The key point is, the contribution of external pressure to enthalpy is the integral of the equation of state of each material on the pressure-axis, and the difference between the integral of product and reactant is the contribution of external pressure to the enthalpy change of the reaction (Scheme 1, [4]). Second, dynamically, the reaction has strong topochemical features, and the relative positions of the building blocks in the crystal structure of the reactant affect the reaction significantly. Here researchers define “topochemical reaction” as a solid-state reaction with the crystal structure of product similar to that of the reactant, and the atoms in the crystal structure of the product can be clearly related to those in the reactant.
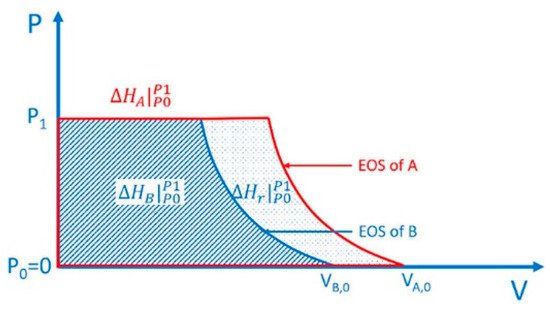
Scheme 1. Contribution of external pressure to the enthalpy of a substance equals to the integral of its equation of state on the pressure-axis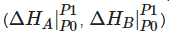
and the contribution to the enthalpy change of a reaction (A = B) equals to the difference of the integrals
Reprinted with permission from ref. [4]. Copyright 2021 American Chemical Society.
2. PIP: Addition Reactions
2.1. Alkyne and Acetylides
As the simplest alkyne, acetylene (C2H2) is an excellent model for the research of PIP. The addition polymerization of acetylene at AP was widely investigated in the last century and Nobel Prize was awarded to the research on conductive polyacetylene (PA). The PIP of acetylene has completely different reaction routes to those under AP. Under HP, acetylene solidifies into crystal and starts to polymerize at 3.5 GPa at room temperature, with the main product confirmed as trans-PA [10][11][12][13], whereas, at 77 K, solid acetylene polymerizes at 12 GPa, consequently forming cis-PA [14]. Since it is well known that the trans-PA is more stable, researchers need to clarify the reaction mechanism of this selective PIP of acetylene. Researchers used Paris-Edinburgh Press (PE press) as a HP generation apparatus to avoid the photoactivation and facilitates the in situ neutron diffraction [15][16][17]. The crystal structure of deuterated acetylene at the critical reaction pressure of 5.7 GPa was determined by in situ neutron diffraction, from which researchers found the possible reaction route to produce cis-PA [18]. As shown Figure 1a (left), the previous proposed bonding route along a+b or a−b direction will result in trans-PA (grey line route) [12][13], while there is another bonding route along the a+c and a−c direction with DC of 3.1 Å (yellow line), which will promote the formation of cis-polymer. The fact that anisotropic thermal factors of carbon and hydrogen (deuterium) reach maximum in the a−c plane also supports this bonding route. On the other hand, in the metadynamics simulation, it also shows that the reaction also takes place in the a+c direction, and the formation of cis-PA is reproduced (Figure 1a (right)). Additionally, the simulation predicted the production of graphane in a further step of PIP, which was demonstrated by researchers' subsequential experiment. This is a typical example that shows the sp-sp2-sp3 sequence in the PIP, and also reveals the neighbor-first topochemical rule in understanding the PIP process (Figure 1b).
Figure 1. (a) (left) Crystal structure of acetylene at 5.7 GPa. The brown and light blue ellipsoids are carbon and deuterium atoms, respectively. The yellow lines indicate the forming of cis-PA while the grey for trans-PA. (right) Metadynamics simulation results of acetylene under 15 GPa. Reprinted with permission from ref. [18]. Copyright 2017 Angewandte Chemie International Edition; (b) PIP of acetylene, via cis-PA to graphane. Hydrogen is omitted for clarity.
The acetylide anions such as C≡C2− and HC≡C− were also predicted to polymerize into conjugated extended carbon framework with excellent electrical conductivity under HP [19][20][21][22][23][24]. As the simplest acetylides, CaC2 and Li2C2 were investigated under HP [22][25][26][27][28], and researchers firstly evidenced their PIP experimentally, which resulted in a remarkable conductivity enhancement,107-fold for CaC2, and 109-fold for Li2C2 [26][27]. After releasing to the atmospheric pressure, due to the extended covalent conjugated C-C bonding, the high conductivity of the recovered products maintains.
Researchers also monitored the crystal structure variation under HP by in situ diffraction and complementary theoretical optimization. CaC2 crystallizes in a tetragonal phase at ambient and low pressure, and distorts into a monoclinic phase above ~10 GPa. This accelerates the approaching of neighbored C22−, and researchers estimate that the DC is ~2.9 Å at around 20 GPa [26]. Above 20 GPa at room temperature, diffraction cannot provide enough structural details since CaC2 polymerizes into amorphous phase. High resolution gas chromatography mass spectrometry (HR GC-MS) analysis of the hydrated products of the recovered sample provided solid evidence of oligomerization of acetylide. Advanced hydrocarbons were observed besides acetylene. Most of components have the C:H ratio of around 1:1 (Figure 2a), which indicates no obvious disproportionation in the PIP. The slight deviation from 1:1 is likely due to the termination of the free radicals, which was discussed in detail in the work of NaC2H [29]. More importantly, benzene (C6H6) was identified accordingly to HR-GC-MS data. This consists with the reaction sequence of forming chain-belt-sheet as predicted by metadynamics simulation (Figure 2b).
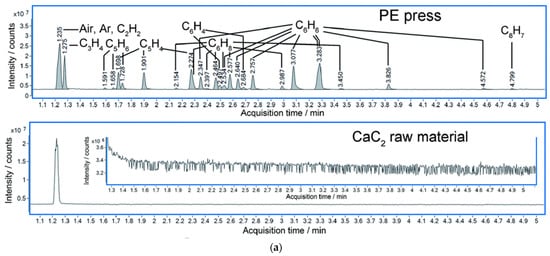
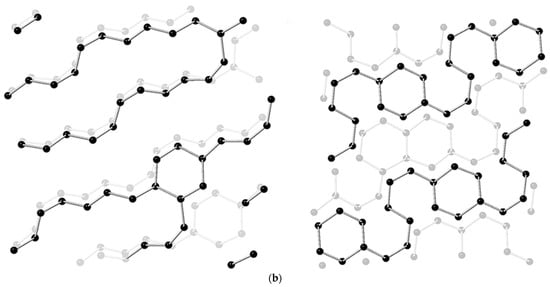
Figure 2. (a) Total ion chromatograms (TIC) of the product of CaC2 (synthesized by PE press) recovered from 26 GPa and CaC2 raw material. (The hydrolysis of the polymerized Cmx− anions in the recovered sample produced C2nH2n according to the chemical reaction equation: CanC2n + 2nH2O = nCa(OH)2 + C2nH2n); (b) Simulated chain and ribbon model of CaC2 at 30 GPa by metadynamics [26].
By comparing the experimental result of Li2C2 under high pressure with the theoretical calculated IR spectra, the carbon-ribbon structure in the Li2C2 polymeric product was identified, and many advanced hydrocarbons in the hydrated products of the recovered sample were detected by GC-MS, which also supports the addition PIP of C22−, and the ribbon structures (Figure 3a) [27]. However, different from CaC2, there were a portion of products with the C:H ratio significantly deviating from 1:1, which revealed the occurrence of the disproportionation reaction. Compared with the Ca2+ in CaC2, the Li+ cation is more active to diffuse. This leads to disproportion and composition segregation under compression, promoting the formation of Li-rich and C-rich phases. In HPHT experiments, crystalline phase Li3C4 with zigzag carbon-ribbon structure (lithium polyacenide) and LiC2 with intercalation structure (lithium graphenide) were indeed obtained (Figure 3b) [28]. Researchers also predicted a series of lithium polycarbides with a stoichiometric ratio of Lin+1C2n (Figure 3c), which share the common feature of zigzag nano-ribbon structure with various width, and LiC2 (n = ∞) and Li3C4 (n = 2) are the two terminal compounds (Figure 3b) [28].
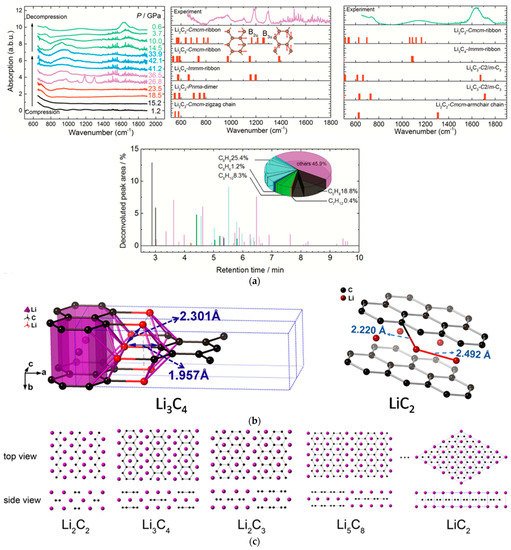
Figure 3. (a) The IR spectra of the Li2C2 (experiment) and the predicted Li-C phases (simulation) under external pressure (above); The relative area of the deconvoluted peaks of the hydrocarbons in the hydrolyzed product (below) in the GC-MS results. The components with a score lower than 90 are counted in “others”. Reprinted with permission from ref. [27]. Copyright 2017 American Chemical Society; (b) The crystal structures of Li3C4 (left) and LiC2 (right); (c) The predicted crystal structures of Lin+1C2n (n = 1, 2, …). The black spheres: C. The purple spheres: Li. Reprinted with permission from ref. [28]. Copyright 2018 American Chemical Society.
As mentioned, acetylene normally reacts at around 5.0 GPa [18], while the polymerization pressures of disubstituted metal acetylides are mostly over 20 GPa (~22 GPa for CaC2, and 35 GPa for Li2C2 [26][27]) due to the repulsion of the electrostatic interaction and spatial blocking of the cations. Researchers also studied the monosubstituted acetylene sodium (NaC2H) with only one charge on the carbon group [29]. By the method of in situ spectroscopy and X-ray diffraction (XRD) technology, PIP of NaC2H was proved beginning from 14 GPa, which is between that of acetylene and acetylides. According to the crystal structure of NaC2H at 11 GPa (the maximum pressure at which researchers can still have usable diffraction data, Figure 4a (left), the DC is around 3.0 Å (d3), and by extrapolation of the d-P curve (Figure 4a (right)) to 14 GPa, the DC was around 2.9 Å. This is similar to that of CaC2 [26]. The specific reaction is shown in Figure 4b. This is a free radical addition process. Due to the produced species needing to absorb or lose at most two hydrogen atoms for pairing their two radicals, the hydrolysis products always have a C:H ratio within CxHx±2 (Figure 4b).
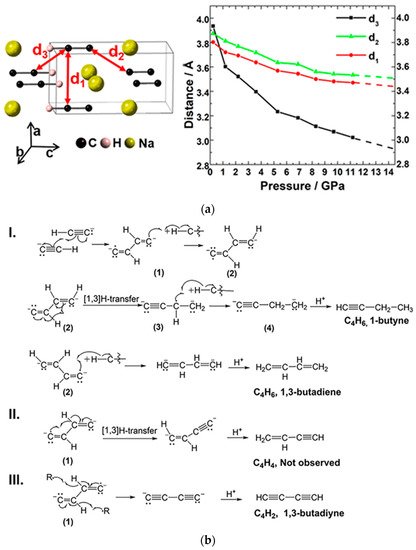
Figure 4. (a) Crystal structure of NaC2H and the evolution of d1, d2, and d3 as a function of pressure. d1, d2, and d3 represent different intermolecular C···C distance; (b) the reaction routes of the NaC2H dimerization and the following H-transfer process. Reprinted with permission from ref. [29]. Copyright 2019 American Chemical Society.
The PIP of C≡C groups should be the simplest PIP reactions. The reactions are likely to follow a free radical route. Though the HP environment prevents us to trap or detect the radicals, the hydrated products of the recovered polyacetylide sample CnHn+2, and CnHn−2 etc., provides indirect evidence. The reactions seem to follow the neighbor-first rule, which is a feature of topochemical route. Regardless of the reaction pressure, the bonding share approximately the same DC, ~3.0 Å, which is a good reference for other unsaturated groups. The hybridization of carbon in PIP follows a sp-sp2-sp3 sequence. The PIP can proceed to the end, or stop at half-way, depending on the reactivity of the unsaturated groups and the pressure applied, which provides more controlling factors for synthesizing advanced materials.
2.2. Aromatic Compounds
As important building blocks for PIP, aromatic compounds also attract continuously attentions. Since the 1980s, benzene (C6H6), as the proto model of aromatic systems, has been systematically explored in the high-pressure research [30][31][32][33][34][35][36][37][38][39][40]. In recent years, through optimizing the synthetic conditions (a slow compression/decompression rate), one-dimensional sp3 carbon nanothreads were identified from the sample recovered from compressing C6H6 at room temperature [41][42]. Compared with the traditional sp2 nanotube, higher strength and stiffness were expected for the diamond-type nanothreads. Similar results were observed in other aromatics cases, such as pyridine (C5H5N) [43][44], thiophene (C4H4S) [45], furan (C4H4O) [46], and aniline (C6H5NH2) [47][48]. Several reaction routes, including [4+2] addition and 1-1′ addition/coupling, were investigated for benzene, and many models were proposed for the nanothreads [49][50][51]. However, probably due to the diversity of the local structures in the solid sample before reaction, there is still existing considerable confusion about the reaction pathway and mechanism.
To investigate a model compound with well-defined ordered structure (“monodispersed” local structure), researchers explored the 1:1 benzene (C6H6) and hexafluorobenzene (C6F6) cocrystal [52][53]. It has a strong electrostatic attraction between the two components, which leads to the C6H6-C6F6 rings being alternately stacked in column regularly [52]. After releasing from 20 GPa, PIP consequently resulted in a layered structure of H/F substituted graphane. Before reaction occurrence, the structure maintained the column stacking, the column was remarkably titled, and, in fact, the molecules formed closed packed layers (Figure 5a). The DC between C6H6 and C6F6 at 20 GPa were decreased to approximately 2.8 Å at room temperature (without thermal correction), reaching the critical reaction distance of previous benzene molecules (2.8~3.0 Å [54]). However, since there are several intermolecular C-C distances closed to DC, a [4+2] reaction is probably preferred according to the neighbor-first rule, but is not exclusive (Figure 5b).
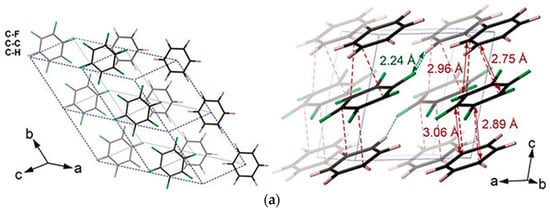
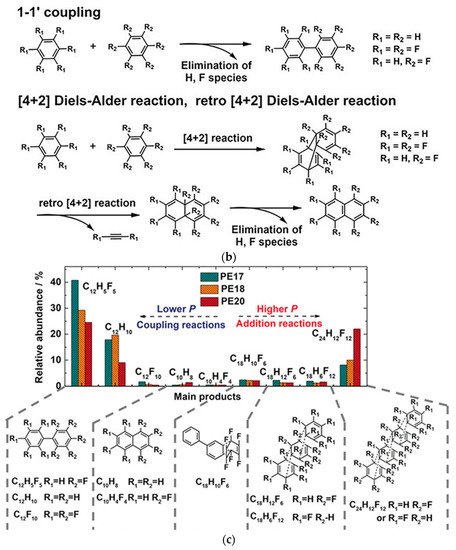
Figure 5. (a) Crystal structure of C6D6-C6F6 before reaction (20 GPa): (left) viewed perpendicular to (102) and (right) approximately along. Reprinted with permission from ref. [55]. Copyright 2020 Nature; (b) Selected elemental reactions of the CHCF cocrystal under high pressure; (c) A histogram of the comparison between the relative abundance of major components in different high-pressure samples from GC-MS results. Reprinted with permission from ref. [53]. Copyright 2019 Angewandte Chemie International Edition.
Then, researchers used HR GC-MS to investigate the reaction intermediates to find the primary reaction models in PIP. Taking the standard mass library and the retention time of commercial samples as reference, researchers concluded that the elemental reactions include Diels-Alder, retro-Diels Alder, and 1-1′ coupling reactions (Figure 5b). By comparing the compositional contents of products recovered from different pressures, researchers found that product content from the [4+2] Diels-Alder addition route was increased under higher pressure (Figure 5c), rather than the coupling reaction, which is also supported by spectroscopic analysis. Unexpectedly, researchers did not observe the 1-D nanothread reported in the PIP of benzene, etc., but found an ordered 2-D H/F substituted graphane structure.
After researchers' work, more similar aromatic cocrystal systems were recently reported, including two polycylic 1:1 arene-perfluoroarene cocrystals, naphthalene/octafluorona-phthalene (NOFN), and anthrancene/octafluoronaphthalene (AOFN) [56][57]. Owing to the strong intermolecular reactions between the larger conjugation, their π-π stacked column structure was more stable under compression, and the molecules only tilted slightly with respect to the stacking direction. The DC was along the interplanar centroid-centroid line of polycylic rings, determined as around 2.8 Å for NOFN and 2.7 Å for AOFN, and similar to that of reported aromatics. In contrast, the PIP products are one-dimensional polymers for both NOFN and AOFN.
Besides benzene-based derivates, heterocyclic compounds, especially nitrogen-containing cyclic molecules, also attract great scientific attentions [58][59][60]. Researchers investigated the structural evolution and chemical reaction processes of 1H-tetrazole (H2CN4) [60], a nitrogen-rich compound with a high nitrogen content of 80%, up to 100 GPa for the purpose of creating a novel polynitrogen material with high energy density. Through the in situ synchrotron XRD, an irreversible reaction was demonstrated in the range of 60 to 100 GPa, which was also evidenced by the strong fluorescence signal detected in the recovered sample in Raman spectroscopy. Intermolecular bonding between the C and N atoms from neighbored molecules, respectively, was proposed, according to theoretical investigation (Figure 6a). However, the bonding of nitrogen seems much harder than that of carbon. No chemical reaction proceeds even when the closest intermolecular C…N reaches 2.52 Å at 48.5 GPa, which was much smaller than the DC of unsaturated carbon groups (Figure 6b). The consequence obviously shows that the DC values are highly elemental dependent, probably due to the different electronegativity and/or bonding formation energy.
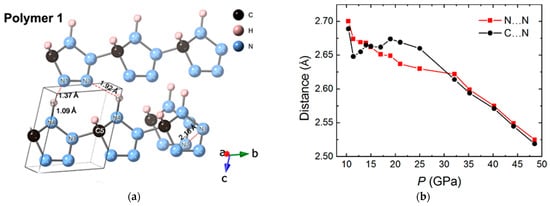
Figure 6. (a) Polymeric structure of 1H-tetrazole under 100 GPa, optimized from the structure obtained from the XRD result at 100.3 GPa; (b) Closet intermolecular C…N and N…N distance as a function of pressure. Reprinted with permission from ref. [60]. Copyright 2021 Royal Society of Chemistry (RSC) Publishing.
From researchers' research, researchers can demonstrate that the PIP of aromatics has various elemental reactions, which can coexist and compete mutually. The intermolecular distances are not far from each other, so the neighbor-first rule may not dominate. By modifying the synthetic conditions, the product may change, which brings great challenges, but also great opportunities.
2.3. Alkynlphenyl Systems
Generally, the PIP of alkynes proceeds at a relatively lower pressure, while the products often lack long-range ordering at room temperature owing to the thermal effect. In contrast, for the aromatics, the products often have better ordering, but the pressures required are much higher. To synthesize ordered polymeric materials via topochemical PIP at relatively low pressure, researchers investigated the PIP of 1,4-diphenylbutadiyne (DPB), aiming to take the advantage of both aryl and ethynyl [61]. Researchers obtained crystalline armchair graphitic nanoribbons, and, importantly, revealed a new PIP route, dehydro-Diels−Alder (DDA) reaction, which conforms exactly to the topochemical strategy. Figure 7a displays the in situ XRD patterns under HP. Before chemical reactions, DPB maintained the low-pressure structure (monoclinic phase). The crystal structure at the reaction threshold pressure of 10 GPa showed that the intermolecular distance 3.22 Å (between the alkynyl and phenyl group) and 3.24 Å (between phenyl groups) were both much smaller than those between diynes, where the shortest distance is 4.29 Å, and is still greater than the commonly required distance (4 Å) for 1,4-addition polymerization of diynes at ambient conditions (Figure 7b). It is the aryl groups that provide a large steric hindrance to prevent the direct addition reaction of triple bonds from approaching and bonding, and hence avoid the possible 1,4-addition product. In such a local structure, the new reaction path, [4+2] DDA reaction between phenyl and alkyne groups, is activated (Figure 7c), and results in the formation of a graphitic ribbon structure.
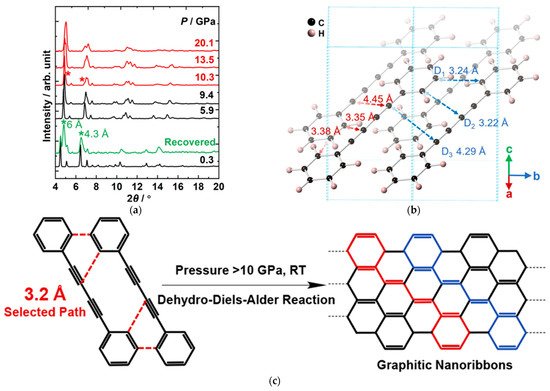
Figure 7. (a) Selected in situ XRD of DPB under high pressure. The peaks from the PIP-DPB are marked by the asterisks; (b) Crystal structure of DPB optimized by density functional theory (DFT) calculation based on the experimental lattice under 10 GPa. (c) Proposed DDA reaction. Reprinted with permission from ref. [61]. Copyright 2020 American Chemical Society.
Subsequently, a similar DDA reaction was also observed for 1,3,5-triethynylbenzene (TEB), where three ethynyl groups were located in the meta-position of a phenyl ring [62]. Under compression, a one-dimensional nanoribbon structure containing sp, sp2, and sp3 carbons was obtained (Figure 8a). Through in situ spectroscopic analysis, researchers found that the phenyl moiety and the ethynyl groups reacted synergistically at a low pressure of 4 GPa. As displayed in Figure 8b, the crystal structure at 3.6 GPa determined by in situ XRD revealed that the DC for PIP was around 3.3 Å, longer than that of aromatics (2.8 Å) and even alkynes (3.1 Å), but close to the distance of the forementioned DPB case (3.24 Å). Accompanied by the structure by Rietveld refinement and NMR experimental data, the theoretical simulation of PIP process proposed reasonable product models, most of which were formed via the intermolecular [4+2] DDA reaction route between the ethynylphenyl groups and the phenyl ring (Figure 8c). Although the original location of the involved ethynyl groups is relatively far from the benzene ring (d2 = 3.89 Å and d7 = 4.10 Å), the tendency of forming six-member rings is still favored, which is supposedly due to the flexibility of the terminal ethynyl, in contrast to the restrained alkynyl moieties in DPB. Moreover, it also could reasonably explain the remarkable decrease in the reaction pressure.
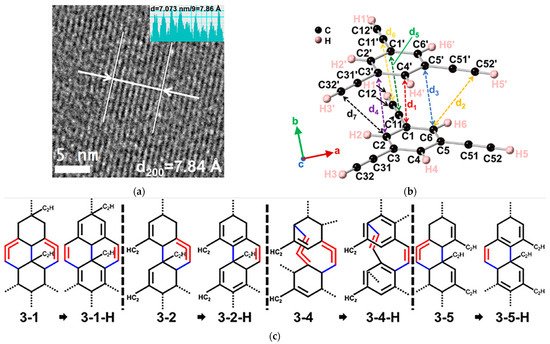
Figure 8. (a) High-resolution transmission electron microscope (TEM) of the recovered sample PE5, which were synthesized at 5 GPa using PE Press; (b) Crystal structure of TEB optimized at 3.6 GPa. The close intermolecular C⋯C distances are d1 (3.39 Å), d2 (3.89 Å), d3,4,5 (3.30 Å), d6 (3.33 Å) and d7 (4.10 Å); (c) Models of ribbons (3-1, 3-1-H, 3-2, 3-2-H, 3-4, 3-4-H, 3-5 and 3-5-H). New bonds are in blue, and double bonds are in red. Reprinted with permission from ref. [62]. Copyright 2021 American Chemical Society.
In summary of the addition polymerization via PIP, researchers can conclude the critical reaction distance rule for the PIP of specific unsaturated functional groups. When approaching a certain distance (~3.0 Å), unsaturated atoms generally tend to bond. The critical distance rule greatly helps us to design suitable precursors for synthesizing novel carbon materials via PIP. As presented Table 1, there are still several factors influencing the critical distance, including the functional groups, reaction element types, and electron charge numbers. The DC of pure aromatics (2.8 Å) is relatively shorter than that of alkynes (3.1 Å), which means that more compression is required to initiate the reaction. Aromatics always have extra stability but worse reactivity. When the ethynyl groups are introduced into a benzene ring, the delocalized conjugation is expanded spatially, and the benzene ring is “activated” by the ethynyl. When the local structure allows, a synergistical reaction [4+2] is preferred with longer DC. It seems that the synergistical bonding naturally has longer DC, since the two bonding can promote each other. In addition, comparing DPB with TEB, the latter molecule has a terminal ethynyl group, and the DC is accordingly increased, probably since the terminal ethynyl has more dynamic freedom, while for the metal carbide, as the charge of C22− caused strong electrostatic repulsion, it is harder to be activated, and a smaller DC is required for triggering reactions.
Table 1. Intermolecular distance threshold for PIP of unsaturated compounds.
| Compounds | DC (Å) | Reaction at Initiation | Pressure (GPa) |
|---|---|---|---|
| Acetylene (C2H2) | 3.1 [18] | free radical reaction | 5.7 |
| Sodium Monoacetylene (NaHC2) | 2.9 [29] | 11 | |
| Calcium Acetylene (CaC2) | 2.9 [26] | 20 | |
| Lithium Acetylene (Li2C2) | - | 33 | |
| Benzene (C6H6) | 2.8 [54] | Diels-Alder/1-1′ coupling, etc. reactions |
18 |
| Benzene-hexafluorobenzene (C6H6-C6F6) |
2.8 [53] | 20 | |
| 1,4-diphenylbutadiyne (DPB) | 3.2 [61] | Diels-Alder reaction | 10 |
| 1,3,5-Triethynylbenzene (TEB) | 3.3 [62] | 4 |
3. PIP: Condensation Reaction with Hydrogen Transfer
Condensation means the production of polymers together with small molecules. Roughly speaking, this is not preferred, similar to the addition reactions under HP, since the volume shrinkage is not significant. However, some pressure-induced condensation polymerizations are often reported. For example, the hydrocarbons are often reported to transform to graphite or diamond, with a loss of hydrogen [63][64][65][66]. This is a decomposition process in the view of composition, but also a condensation polymerization process in the view of structure. At times, hydrogen may leak from the HP sample environment to the gasket material or ambient environment, which promotes the condensation process in a thermodynamical view, and was discussed in researchers' recent work [4]. Owing to the excellent dynamics of hydrogen under HP, the transfer and elimination of hydrogen-bearing molecules is likely the most important step in the condensation polymerization under HP.
3.1. Alkane
Alkane is highly inert and difficult to employ for polymerization since all the atoms are saturated by the C-H sigma bond. However, under natural conditions, the synthesis of heavy hydrocarbons or hydrogen-carbon polymers could occur under HPHT conditions without a catalyst on a planet or on Earth. Experimentally, by using the laser-heating diamond anvil cells (DAC) up to 2 GPa and 1000–1500 K, methane (CH4) was demonstrated to produce advanced alkanes (C2~C4), molecular hydrogen, unsaturated hydrocarbon, and graphite [63]. Moreover, methane decomposes to form diamonds at 10–50 GPa and 2000–3000 K [64]. At 48 GPa, methane dissociates at 1200–1500 K with the formation of hydrogen, and it decomposes further to form large amounts of solid carbon, hydrogen, and higher hydrocarbons when heating above 1500 K [65]. From the view of the reaction mechanism, the removal of hydrogen atom from alkane was the initial step under HPHT, and the remaining carbon-rich part tended to link together and then form larger molecules with thermodynamical stability. This process could be regarded as the polymerization of alkane, and the fundamental principle is the condensation reaction with the elimination of molecular hydrogen (H2).
Recently, researchers explored n-hexane and cyclohexane under extreme conditions. Under high pressure and room temperature, both n-hexane and cyclohexane only underwent reversible phase transitions up to approximately 42 GPa without any chemical reaction [66]. By using resistive heating combined with DAC, the dehydrogenation reactions and polymerization under HPHT proceed for both n-hexane and cyclohexane. For n-hexane under 21.4 GPa, when heated to 745 K, intense fluorescence appeared, indicating the formation of unsaturated compound with conjugated structures such as aromatics or conjugated alkenes. At 932 K, new alkanes were formed as new peaks appearing in the region of sp3-CH. At 995 K, an extended carbon structure was formed and determined to be a partially hydrogenated graphite (Figure 9a). The whole process was the dehydrogenation of alkanes, formation of conjugated hydrocarbons, and subsequently the production of polymeric carbon-based species. For cyclohexane, unlike n-hexane, no strong fluorescence was observed during the heating process, indicating a different reaction pathway, which still needs further investigation, whereas the final products synthesized from cyclohexane were very similar to that of n-hexane, which means the hydrogen transfer reaction and condensation process also dominated under the HPHT conditions [66]. Some reported results of alkanes under HPHT are summarized in Figure 9b.
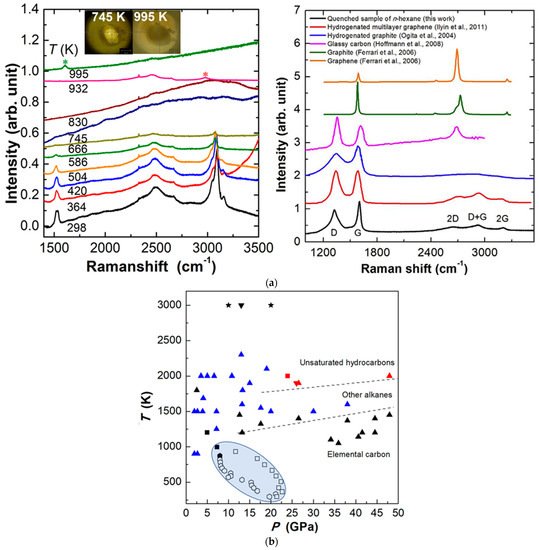
Figure 9. (a) Selected Raman spectra of n-hexane with increasing temperature at 21.4 GPa. The new peaks are marked by the asterisks (left); Raman spectra of quenched sample outside DAC (right); (b) C-H system experiment results under HPHT with different staring materials. Reprinted with permission from ref. [66]. Copyright 2021 Elsevier.
3.2. Nitriles
The condensation polymerization via hydrogen transfer process was also observed under HPRT conditions. Acetonitrile (CH3CN) is a prototype of nitriles, and widely used as a reaction solvent for its stability. Its C-H bond is too stable to be activated. However, in researchers' study, the C-H bond of acetonitrile was activated by cyano groups at high pressure and room temperature [67]. In situ mid-IR spectra of acetonitrile demonstrated that the C=N/C=C and amino groups emerged at 23 GPa and maintained when releasing to ambient pressure (Figure 10a,b). This clearly indicated that the cyano groups underwent a hydrogen transfer process, from CH3 to C≡N along the CH∙∙∙N hydrogen bond, and generated amino groups. Similar phenomena have also been reported in hydrogen cyanide under high pressure [68].
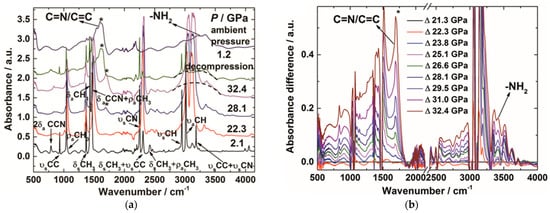
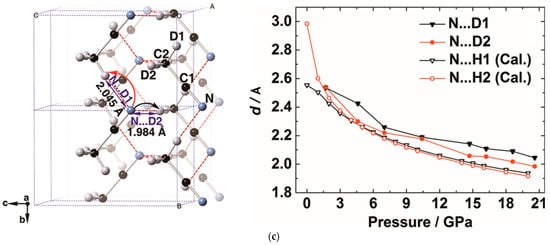
Figure 10. (a) Selected in situ IR patterns of CH3CN at different pressures. The asterisks display the presence of C=N/C=C group, indicating the PIP of CH3CN; (b) Selected IR spectra subtracted by the pattern of 21.3 GPa. The increasing absorbance of the C=N/C=C group is marked by the asterisks; (c) The crystal structure of CD3CN at 20.6 GPa and the evolution of selected intermolecular atomic distances as a function of pressure. N···D1 and N···D2 represent the corresponding intermolecular atomic distances obtained from the neutron diffraction data; N···H1 (Cal.) and N···H2 (Cal.) represent the distance obtained from DFT calculations. Reprinted with permission from ref. [67]. Copyright 2016 Angewandte Chemie International Edition.
This reaction route was clearly uncovered by in situ neutron powder diffraction, by which researchers determined the crystal structures at the reaction threshold pressure of 20.6 GPa. The intermolecular distances between N∙∙∙H(D) are compressed to 2.0 Å, which was consistent with the density functional theory (DFT) calculation results (Figure 10c). This DC is significantly shorter than the sum of van der Waals radii (2.75 Å) of H and N by ~27%. Since the N-H bond length was approximately 1 Å and the H atom only needs to move 1 Å to transfer, considering the thermal vibration and the tunneling effect, researchers concluded that under HP the hydrogen transfer from -CH3 to -CN along the hydrogen bond, as indicated by the arrows in Figure 10c.
In contrast to the alkanes, acetonitrile is unsaturated. During the hydrogen transfer process, active intermediates (possibly similar to CH2=C=NH and CH≡C-NH2) was produced and the polymerization reaction (C-C bonding) was triggered simultaneously. This is thermodynamically preferred, and promoted the hydrogen transfer process. By using DAC as high-pressure generation apparatus, researchers obtained a yellow product containing sp2-, sp3-carbon, imine (-CH-C=NH), and amino groups(-NH2). When using PE press which has a relatively larger pressure gradient and easily cause cracks in the gasket, an obvious releasing of ammonia gas was detected, and the rest of the black product was found to be a graphitic polymer. Therefore, the whole polymerization process leading to the graphitic polymer can be regarded as a consecutive condensation reaction, accompanied with the loss of NH3. Similar condensation polymerizations were also reported in other nitrogen-containing molecules, for instance octahydro-1,3,5,7-tetranitro-1,3,5,7-tetrazocine (HMX) which has nitro groups, with the high temperature condition needed for activation [69].
With respect to the reason of small molecule loss, researchers conclude the pressure gradient is a significant factor to accelerate hydrogen transfer dynamically, which has been observed in several systems experimentally and discussed theoretically. For example, in the forementioned acetylene and acetonitrile, when the sample was not well sealed or obvious gasket breaking appeared, a more conjugated graphene-like structure was formed in the “unsuccessful” experiments. For the HPHT experiments in alkanes, heating will also easily result in some cracks in the gaskets. In these situations, a large pressure gradient would be formed, which could promote the leak of small molecules produced from the hydrogen transfer reaction. As the reaction proceeds, the elimination of small molecules would conversely promote the hydrogen transfer reaction and lead to the formation of an extended carbon-base framework under high pressure.
References
- Akaishi, M.; Kanda, H.; Yamaoka, S. Synthesis of diamond from graphite-carbonate system under very high temperature and pressure. J. Cryst. Growth 1990, 104, 578–581.
- Irifune, T.; Kurio, A.; Sakamoto, S.; Inoue, T.; Sumiya, H.; Funakoshi, K.-I. Formation of pure polycrystalline diamond by direct conversion of graphite at high pressure and high temperature. Phys. Earth Planet. Inter. 2004, 143, 593–600.
- Isobe, F.; Ohfuji, H.; Sumiya, H.; Irifune, T. Nanolayered diamond sintered compact obtained by direct conversion from highly oriented graphite under high pressure and high temperature. J. Nanomater. 2013, 2013, 380165.
- Wang, Y.; Yang, X.; Tang, X.; Wang, X.; Li, Y.; Lin, X.; Dong, X.; Yang, D.; Zheng, H.; Li, K. Pressure gradient squeezing hydrogen out of MnOOH: Thermodynamics and electrochemistry. J. Phys. Chem. Lett. 2021, 12, 10893–10898.
- Schettino, V.; Bini, R. Molecules under extreme conditions: Chemical reactions at high pressure. Phys. Chem. Chem. Phys. 2003, 5, 1951–1965.
- Schettino, V.; Bini, R. Constraining molecules at the closest approach: Chemistry at high pressure. Chem. Soc. Rev. 2007, 36, 869–880.
- Yang, X.; Wang, X.; Wang, Y.; Li, K.; Zheng, H. From molecules to carbon materials-High pressure induced polymerization and bonding mechanisms of unsaturated compounds. Crystals 2019, 9, 490.
- Yoo, C.-S. Chemistry under extreme conditions: Pressure evolution of chemical bonding and structure in dense solids. Matter Radiat. Extrem. 2020, 5, 018202.
- Wang, X.; Li, K.; Zheng, H.; Zhang, P. Chemical reactions of molecules under high pressure. Chem. Bull. 2019, 82, 387–398.
- Aoki, K.; Kakudate, Y.; Yoshida, M.; Usuba, S.; Tanaka, K.; Fujiwara, S. Raman scattering observations of phase transitions and polymerizations in acetylene at high pressure. Solid State Commun. 1987, 64, 1329–1331.
- Aoki, K.; Kakudate, Y.; Usuba, S.; Yoshida, M.; Tanaka, K.; Fujiwara, S. High-pressure Raman study of liquid and crystalline C2H2. J. Chem. Phys. 1988, 88, 4565–4568.
- Aoki, K.; Usuba, S.; Yoshida, M.; Kakudate, Y.; Tanaka, K.; Fujiwara, S. Raman study of the solid-state polymerization of acetylene at high pressure. J. Chem. Phys. 1988, 89, 529–534.
- Aoki, K.; Kakudate, Y.; Yoshida, M.; Usuba, S.; Tanaka, K.; Fujiwara, S. Solid-state polymerization of acetylene under pressure. Synth. Met. 1989, 28, D91–D98.
- Trout, C.C.; Badding, J. Solid state polymerization of acetylene at high pressure and low temperature. J. Phys. Chem. A 2000, 104, 8142–8145.
- Marshall, W.G.; Francis, D.J. Attainment of near-hydrostatic compression conditions using the Paris–Edinburgh cell. J. Appl. Crystallogr. 2002, 35, 122–125.
- Klotz, S.; Besson, J.M.; Hamel, G.; Nelmes, R.J.; Loveday, J.S.; Marshall, W.G. High pressure neutron diffraction using the paris-edinburgh cell: Experimental possibilities and future prospects. High Press. Res. 1996, 14, 249–255.
- Besson, J.; Nelmes, R. New developments in neutron-scattering methods under high pressure with the Paris-Edinburgh cells. Phys. B Condens. Matter 1995, 213, 31–36.
- Sun, J.; Dong, X.; Wang, Y.; Li, K.; Zheng, H.; Wang, L.; Cody, G.D.; Tulk, C.A.; Molaison, J.J.; Lin, X. Pressure-induced polymerization of acetylene: Structure-directed stereoselectivity and a possible route to graphane. Angew. Chem. Int. Ed. 2017, 129, 6653–6657.
- Chen, X.-Q.; Fu, C.; Franchini, C. Polymeric forms of carbon in dense lithium carbide. J. Phys. Condens. Matter 2010, 22, 292201.
- Benson, D.; Li, Y.; Luo, W.; Ahuja, R.; Svensson, G.; Haussermann, U. Lithium and calcium carbides with polymeric carbon structures. Inorg. Chem. 2013, 52, 6402–6406.
- Li, Y.-L.; Luo, W.; Zeng, Z.; Lin, H.-Q.; Mao, H.-k.; Ahuja, R. Pressure-induced superconductivity in CaC2. Proc. Natl. Acad. Sci. USA 2013, 110, 9289–9294.
- Li, Y.-L.; Wang, S.-N.; Oganov, A.R.; Gou, H.; Smith, J.S.; Strobel, T.A. Investigation of exotic stable calcium carbides using theory and experiment. Nat. Commun. 2015, 6, 6974.
- Lin, Y.; Strobel, T.A.; Cohen, R. Structural diversity in lithium carbides. Phys. Rev. B 2015, 92, 214106.
- Efthimiopoulos, I.; Kunc, K.; Vazhenin, G.; Stavrou, E.; Syassen, K.; Hanfland, M.; Ruschewitz, U. Structural transformation and vibrational properties of BaC2 at high pressure. Phys. Rev. B 2012, 85, 054105.
- Efthimiopoulos, I.; Benson, D.E.; Konar, S.; Nylén, J.; Svensson, G.; Häussermann, U.; Liebig, S.; Ruschewitz, U.; Vazhenin, G.V.; Loa, I. Structural transformations of Li2C2 at high pressures. Phys. Rev. B 2015, 92, 064111.
- Zheng, H.; Wang, L.; Li, K.; Yang, Y.; Wang, Y.; Wu, J.; Dong, X.; Wang, C.-H.; Tulk, C.A.; Molaison, J.J. Pressure induced polymerization of acetylide anions in CaC2 and 107-fold enhancement of electrical conductivity. Chem. Sci. 2017, 8, 298–304.
- Wang, L.; Dong, X.; Wang, Y.; Zheng, H.; Li, K.; Peng, X.; Mao, H.-k.; Jin, C.; Meng, Y.; Huang, M. Pressure-induced polymerization and disproportionation of Li2C2 accompanied with irreversible conductivity enhancement. J. Phys. Chem. Lett. 2017, 8, 4241–4245.
- Dong, X.; Wang, L.; Li, K.; Zheng, H.; Wang, Y.; Meng, Y.; Shu, H.; Mao, H.-K.; Feng, S.; Jin, C. Tailored synthesis of the narrowest zigzag graphene nanoribbon structure by compressing the lithium acetylide under high temperature. J. Phys. Chem. C 2018, 122, 20506–20512.
- Han, J.; Tang, X.; Wang, Y.; Wang, Y.; Han, Y.; Lin, X.; Dong, X.; Lee, H.H.; Zheng, H.; Li, K. Pressure-induced polymerization of monosodium acetylide: A radical reaction initiated topochemically. J. Phys. Chem. C 2019, 123, 30746–30753.
- Ellenson, W.D.; Nicol, M. Raman spectra of solid benzene under high pressures. J. Chem. Phys. 1974, 61, 1380–1389.
- Thiery, M.; Leger, J. High pressure solid phases of benzene. I. Raman and X-ray studies of C6H6 at 294 K up to 25 GPa. J. Chem. Phys. 1988, 89, 4255–4271.
- Pruzan, P.; Chervin, J.; Thiery, M.; Itie, J.; Besson, J.; Forgerit, J.; Revault, M. Transformation of benzene to a polymer after static pressurization to 30 GPa. J. Chem. Phys. 1990, 92, 6910–6915.
- Cansell, F.; Fabre, D.; Petitet, J.P. Phase transitions and chemical transformations of benzene up to 550 ℃ and 30 GPa. J. Chem. Phys. 1993, 99, 7300–7304.
- Ciabini, L.; Santoro, M.; Bini, R.; Schettino, V. High pressure crystal phases of benzene probed by infrared spectroscopy. J. Chem. Phys. 2001, 115, 3742–3749.
- Ciabini, L.; Santoro, M.; Bini, R.; Schettino, V. High pressure reactivity of solid benzene probed by infrared spectroscopy. J. Chem. Phys. 2002, 116, 2928–2935.
- Ciabini, L.; Santoro, M.; Bini, R.; Schettino, V. High pressure photoinduced ring opening of benzene. Phys. Rev. Lett. 2002, 88, 085505.
- Jackson, B.; Trout, C.; Badding, J. UV Raman analysis of the C: H network formed by compression of benzene. Chem. Mater. 2003, 15, 1820–1824.
- Ciabini, L.; Gorelli, F.A.; Santoro, M.; Bini, R.; Schettino, V.; Mezouar, M. High-pressure and high-temperature equation of state and phase diagram of solid benzene. Phys. Rev. B 2005, 72, 094108.
- Citroni, M.; Bini, R.; Foggi, P.; Schettino, V. Role of excited electronic states in the high-pressure amorphization of benzene. Proc. Natl. Acad. Sci. USA 2008, 105, 7658–7663.
- Wen, X.-D.; Hoffmann, R.; Ashcroft, N. Benzene under high pressure: A story of molecular crystals transforming to saturated networks, with a possible intermediate metallic phase. J. Am. Chem. Soc. 2011, 133, 9023–9035.
- Fitzgibbons, T.C.; Guthrie, M.; Xu, E.-s.; Crespi, V.H.; Davidowski, S.K.; Cody, G.D.; Alem, N.; Badding, J.V. Benzene-derived carbon nanothreads. Nat. Mater. 2015, 14, 43–47.
- Li, X.; Baldini, M.; Wang, T.; Chen, B.; Xu, E.-s.; Vermilyea, B.; Crespi, V.H.; Hoffmann, R.; Molaison, J.J.; Tulk, C.A. Mechanochemical synthesis of carbon nanothread single crystals. J. Am. Chem. Soc. 2017, 139, 16343–16349.
- Li, X.; Wang, T.; Duan, P.; Baldini, M.; Huang, H.T.; Chen, B.; Badding, J.V. Carbon nitride nanothread crystals derived from pyridine. J. Am. Chem. Soc. 2018, 140, 4969–4972.
- Fanetti, S.; Santoro, M.; Alabarse, F.; Enrico, B.; Bini, R. Modulating the H-bond strength by varying the temperature for the high pressure synthesis of nitrogen rich carbon nanothreads. Nanoscale 2020, 12, 5233–5242.
- Biswas, A.; Ward, M.D.; Wang, T.; Zhu, L.; Huang, H.-T.; Badding, J.V.; Crespi, V.H.; Strobel, T.A. Evidence for orientational order in nanothreads derived from thiophene. J. Phys. Chem. Lett. 2019, 10, 7164–7171.
- Huss, S.; Wu, S.; Chen, B.; Wang, T.; Gerthoffer, M.C.; Ryan, D.J.; Smith, S.E.; Crespi, V.H.; Badding, J.V.; Elacqua, E. Scalable synthesis of crystalline one-dimensional carbon nanothreads through modest-pressure polymerization of furan. ACS Nano 2021, 15, 4134–4143.
- Fanetti, S.; Nobrega, M.M.; Teixeira-Neto, E.; Temperini, M.L.; Bini, R. Effect of structural anisotropy in high-pressure reaction of aniline. J. Phys. Chem. C 2018, 122, 29158–29164.
- Nobrega, M.M.; Teixeira-Neto, E.; Cairns, A.B.; Temperini, M.L.; Bini, R. One-dimensional diamondoid polyaniline-like nanothreads from compressed crystal aniline. Chem. Sci. 2018, 9, 254–260.
- Chen, B.; Hoffmann, R.; Ashcroft, N.; Badding, J.; Xu, E.; Crespi, V. Linearly polymerized benzene arrays as intermediates, tracing pathways to carbon nanothreads. J. Am. Chem. Soc. 2015, 137, 14373–14386.
- Duan, P.; Li, X.; Wang, T.; Chen, B.; Juhl, S.J.; Koeplinger, D.; Crespi, V.H.; Badding, J.V.; Schmidt-Rohr, K. The chemical structure of carbon nanothreads analyzed by advanced solid-state NMR. J. Am. Chem. Soc. 2018, 140, 7658–7666.
- Wang, T.; Duan, P.; Xu, E.-S.; Vermilyea, B.; Chen, B.; Li, X.; Badding, J.V.; Schmidt-Rohr, K.; Crespi, V.H. Constraining carbon nanothread structures by experimental and calculated nuclear magnetic resonance spectra. Nano Lett. 2018, 18, 4934–4942.
- Wang, Y.; Wang, L.; Zheng, H.; Li, K.; Andrzejewski, M.; Hattori, T.; Sano-Furukawa, A.; Katrusiak, A.; Meng, Y.; Liao, F. Phase transitions and polymerization of C6H6–C6F6 cocrystal under extreme conditions. J. Phys. Chem. C 2016, 120, 29510–29519.
- Wang, Y.; Dong, X.; Tang, X.; Zheng, H.; Li, K.; Lin, X.; Fang, L.; Sun, G.A.; Chen, X.; Xie, L.; et al. Pressure-induced Diels-Alder reactions in C6H6-C6F6 cocrystal towards graphane structure. Angew. Chem. Int. Ed. 2019, 58, 1468–1473.
- Ciabini, L.; Santoro, M.; Gorelli, F.A.; Bini, R.; Schettino, V.; Raugei, S. Triggering dynamics of the high-pressure benzene amorphization. Nat. Mater. 2007, 6, 39–43.
- Miao, M.; Sun, Y.; Zurek, E.; Lin, H. Chemistry under high pressure. Nat. Rev. Chem. 2020, 4, 508–527.
- Ward, M.D.; Tang, W.S.; Zhu, L.; Popov, D.; Cody, G.D.; Strobel, T.A. Controlled single-crystalline polymerization of C10H8· C10F8 under pressure. Macromolecules 2019, 52, 7557–7563.
- Friedrich, A.; Collings, I.E.; Dziubek, K.F. Pressure-induced polymerization of polycyclic arene-perfluoroarene cocrystals: Single crystal X-ray diffraction studies, reaction kinetics, and design of columnar hydrofluorocarbons. J. Am. Chem. Soc. 2020, 142, 18907–18923.
- Spaulding, D.K.; Weck, G.; Loubeyre, P.; Datchi, F.; Dumas, P.; Hanfland, M. Pressure-induced chemistry in a nitrogen-hydrogen host—Guest structure. Nat. Commun. 2014, 5, 5739.
- Li, W.; Huang, X.; Bao, K.; Zhao, Z.; Huang, Y.; Wang, L.; Wu, G.; Zhou, B.; Duan, D.; Li, F. A novel high-density phase and amorphization of nitrogen-rich 1H-tetrazole (CH2N4) under high pressure. Sci. Rep. 2017, 7, 39249.
- Gao, D.; Tang, X.; Wang, X.; Yang, X.; Zhang, P.; Che, G.; Han, J.; Hattori, T.; Wang, Y.; Dong, X. Phase transition and chemical reactivity of 1H-tetrazole under high pressure up to 100 GPa. Phys. Chem. Chem. Phys. 2021, 23, 19503–19510.
- Zhang, P.; Tang, X.; Wang, Y.; Wang, X.; Gao, D.; Li, Y.; Zheng, H.; Wang, Y.; Wang, X.; Fu, R. Distance-selected topochemical dehydro-Diels–Alder reaction of 1, 4-diphenylbutadiyne toward crystalline graphitic nanoribbons. J. Am. Chem. Soc. 2020, 142, 17662–17669.
- Li, Y.; Tang, X.; Zhang, P.; Wang, Y.; Yang, X.; Wang, X.; Li, K.; Wang, Y.; Wu, N.; Tang, M. Scalable high-pressure synthesis of sp2–sp3 carbon nanoribbon via polymerization of 1, 3, 5-triethynylbenzene. J. Phys. Chem. Lett. 2021, 12, 7140–7145.
- Kolesnikov, A.; Kutcherov, V.G.; Goncharov, A.F. Methane-derived hydrocarbons produced under upper-mantle conditions. Nat. Geosci. 2009, 2, 566–570.
- Benedetti, L.R.; Nguyen, J.H.; Caldwell, W.A.; Liu, H.; Kruger, M.; Jeanloz, R. Dissociation of CH4 at high pressures and temperatures: Diamond formation in giant planet interiors? Science 1999, 286, 100–102.
- Lobanov, S.S.; Chen, P.-N.; Chen, X.-J.; Zha, C.-S.; Litasov, K.D.; Mao, H.-K.; Goncharov, A.F. Carbon precipitation from heavy hydrocarbon fluid in deep planetary interiors. Nat. Commun. 2013, 4, 2446.
- Yang, X.; Li, Y.; Wang, Y.; Zheng, H.; Li, K.; Mao, H.-k. Chemical transformations of n-hexane and cyclohexane under the upper mantle conditions. Geosci. Front. 2021, 12, 1010–1017.
- Zheng, H.; Li, K.; Cody, G.D.; Tulk, C.A.; Dong, X.; Gao, G.; Molaison, J.J.; Liu, Z.; Feygenson, M.; Yang, W. Polymerization of acetonitrile via a hydrogen transfer reaction from CH3 to CN under extreme conditions. Angew. Chem. Int. Ed. 2016, 55, 12040–12044.
- Aoki, K.; Baer, B.; Cynn, H.; Nicol, M. High-pressure Raman study of one-dimensional crystals of the very polar molecule hydrogen cyanide. Phys. Rev. B 1990, 42, 4298.
- Gao, D.; Huang, J.; Lin, X.; Yang, D.; Wang, Y.; Zheng, H. Phase transitions and chemical reactions of octahydro-1, 3, 5, 7-tetranitro-1, 3, 5, 7-tetrazocine under high pressure and high temperature. RSC Adv. 2019, 9, 5825–5833.
More