Acute myeloid leukemia (AML) is a clonal disorder that affects myeloid progenitor cells residing in the bone marrow (BM). This implies altered differentiation with subsequent abnormal proliferation and accumulation of inadequately matured myeloid cells. The detection of leukemic cells moved in the last two decades from immune-phenotyping to polymerase chain reaction (PCR) and real-time quantitative PCR (RT-qPCR). This technique was shown to be reproducible, accurate and highly sensitive for MRD monitoring, with a significant capacity in predicting prognosis, treatment effectiveness and relapse risk. NGS or massively parallel sequencing is a revolutionary method of DNA and RNA sequencing. It is called parallel because it sequences millions of DNA fragments simultaneously. In the early years of its appearance, Next Generation Sequencing (NGS) platforms were used primarily for cancer research purposes. Recently, they are increasingly emerging as irreplaceable diagnostic tools in clinical settings.
- acute myeloid leukemia (AML)
- RT-qPCR
- MDR
- digital droplet PCR (ddPCR)
- next-generation sequencing (NGS)
- metabolomics analysis
- metabolomic profiling
1. Introduction
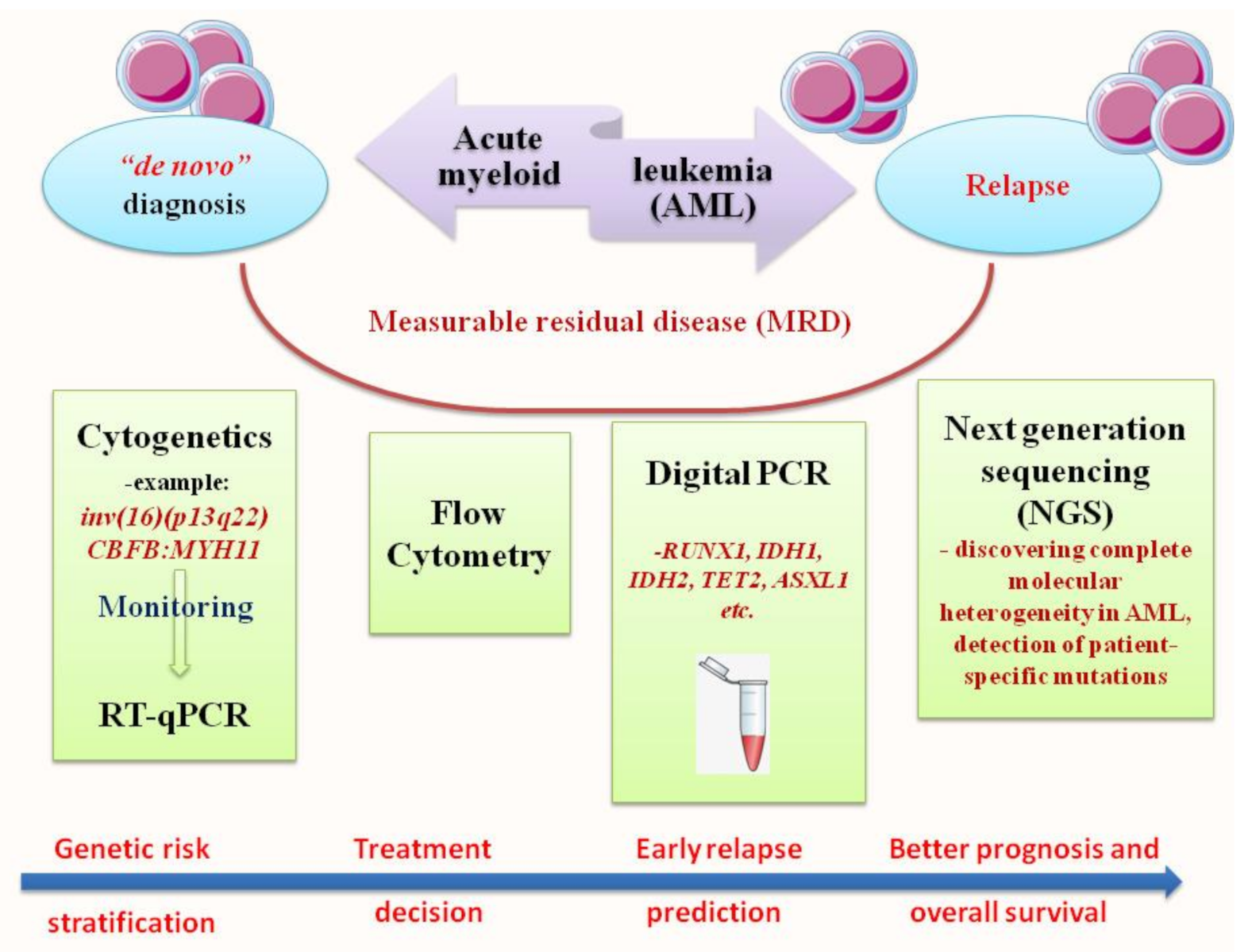
2. RT-qPCR: The Gold Standard for Diagnosis and Prognosis Stratification in AML
3. Next Generation Sequencing (NGS): Unveiling of the Molecular Landscape in Myeloid Neoplasms
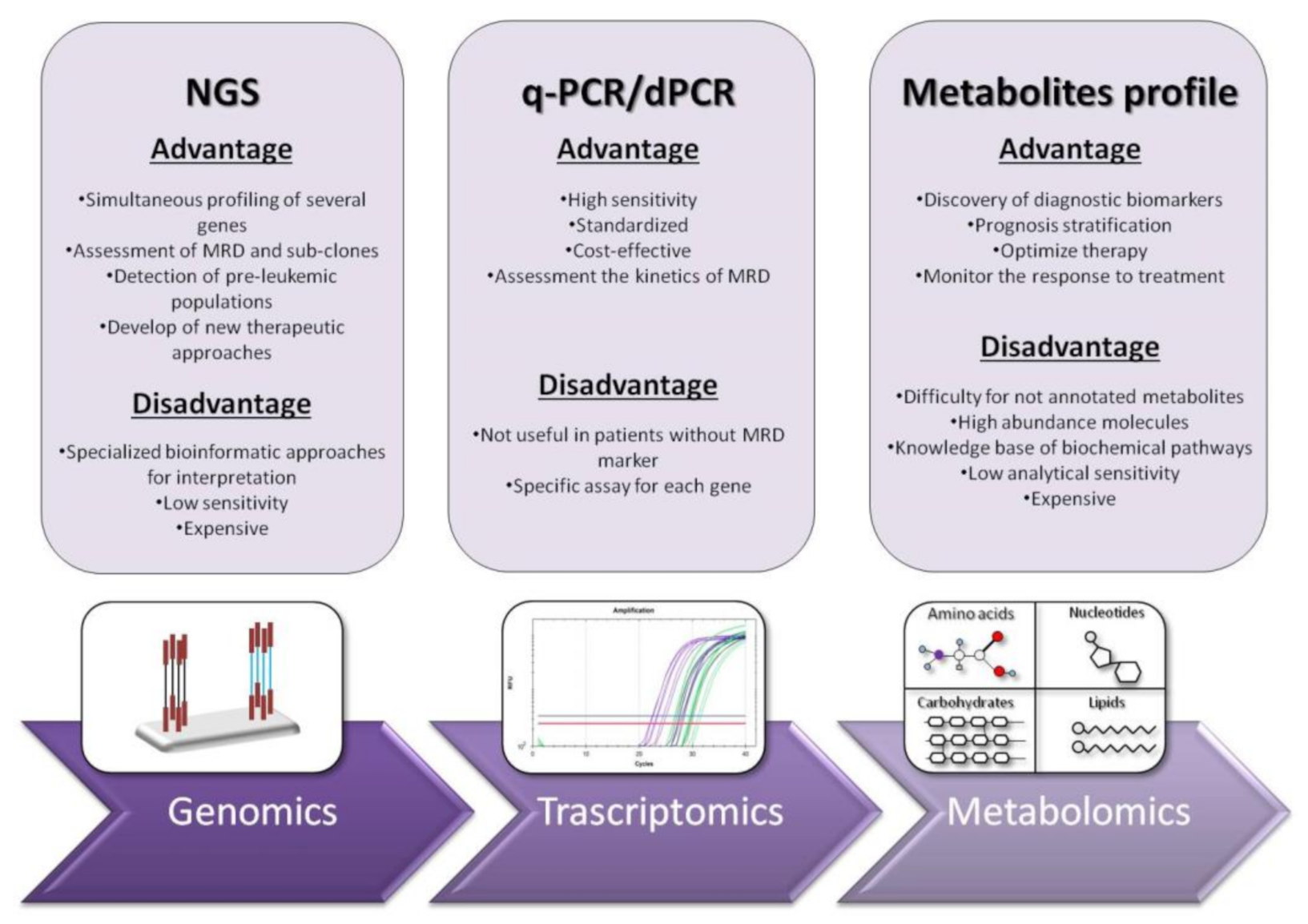
NGS or massively parallel sequencing is a revolutionary method of DNA and RNA sequencing. It is called parallel because it sequences millions of DNA fragments simultaneously. Sequencing may be limited to selected segments of certain genes or to the entire exome [33]. The workflow that NGS runs is as follows: (i) library preparation, (ii) sequencing and (iii) data analysis. In the first stage, the DNA or RNA sample is prepared for sequencing, while fragmenting and adding special adapters to both ends of the fragments. In this manner, the fragments can be amplified. In the next step (ii), the fragments are placed in a flow cell and sequencer. The clusters of DNA or RNA fragments are then amplified in a process called cluster generation, creating millions of copies of single-stranded DNA or RNA. In this very step, chemically modified and fluorescently labeled nucleotides, through the principle of natural complementarity, bind to the DNA template and prevent the next base incorporation. The last step (iii) is data analysis, i.e., the determination of incorporated nucleotides [34]. In the early years of its appearance, NGS platforms were used primarily for cancer research purposes. Recently, they are increasingly emerging as irreplaceable diagnostic tools in clinical settings. So far, there are several commercially available NGS myeloid panels. They target about 30 genes directly or indirectly involved in the pathophysiology of myeloid neoplasms. Depending on their function, these genes may belong to the group of transcription factors, epigenetic modifiers, signal molecules, etc. The clinical use of NGS proved to be important in demystifying myeloid neoplasms that lack classical chromosomal or gene aberrations. For example, in Ph-MPNs, in addition to the presence of “driver” mutations, the application of NGS in a clinical setting reveals new mutations that facilitate risk stratification and treatment decision [35]. NGS reveals an entirely new concept of disease understanding, pushing several layers deeper into the genetic profile and directly opening the possibility for new therapeutic approaches. The co-occurrence of newly discovered and “driver” mutations also offers a new concept regarding prognosis [36]. Of particular interest here is the detection of a mutation that will predict early progression (development of) in secondary AML. Thus, the co-existence of ASXL1, SRSF2, EZH2, IDH1 and IDH2 in PMF patients is associated with shorter leukemic-free survival and an increased risk of leukemic transformation [35][37]. Similarly, in ET and PV patients, mutations in the IDH2, U2AF1, EZH2, TP53, SH2B3 and SF3B1 genes are associated with a worse prognosis [38]. NGS enables more detailed detection of each patient’s molecular map and further efficient selection of HSCT candidates. The idea of MRD assessment with NGS in AML patients has existed since the very beginning of NGS expansion in clinical settings. Scientific studies have shown that 96% of AML patients have at least one driver mutation, and 86% have at least two [39]. By improving clinical applicability and increasing sensitivity, NGS can be a valid tool for MRD assessment in AML patients, especially among those with rare gene mutations. One study, based on mutation detection in NPM1 and FLT3-ITD genes, showed that NGS had assured MRD assessment and 95% concordance with RT-qPCR for mutated NPM1 [40]. In a study by Morita et al. using targeted sequencing of 295 genes in 131 AML patients, the lower cumulative incidence of relapse (CIR) and better overall survival (OS) were found among patients who had no residual mutations until 30 days after induction therapy [41]. RUNX1 gene evaluation with NGS is also a possible choice for MRD analysis in AML patients. In one study in this context, mutational burden <3.61% was associated with better event-free survival (EFS) and OS [42]. In a large study of 482 AML patients using a 54-gene NGS panel, samples were sequenced at the time of diagnosis and in the phase of clinical remission after induction chemotherapy. It was found that, in almost 90% of patients, at least one detectable mutation was present at the time of diagnosis. Using the same assay, the same analysis was performed after therapy, and a mutation was observed in 51% of the patients. A conclusion of great importance in this study is the fact that patients who had only DTA mutations (DNMT3A, TET2 and ASXL1) had a reduced risk of developing relapse, while patients with persistent mutations in other genes had an increased risk of developing relapse [43]. The fact that at least one leukemic mutation is present in a large number of AML patients permits us to believe that any of these mutations may be an appropriate marker for MRD, and NGS can provide an effective MRD assessment. Additionally, NGS can detect reciprocal gene rearrangements such as PML-RARA, RUNX1-RUNX1T1 and CBFB-MYH11. The NGS method is particularly superior in detecting intra-chromosomal rearrangements compared to FISH, which can detect these changes with great sensitivity only on larger chromosomes [44]. However, even if NGS can be used to detect MRD markers, association with cytogenetic and PCR-based approaches are essential to quantify correctly the presence of target [45]. At this time, detection of novel mutations or gene variants by NGS is not associated with change in treatment plan since their functional consequences are not yet fully understood. Furthermore, NGS is considered useful to define relapse, but it would be necessary to identify clinically the meaning of novel genetic mutation and their impact on disease patterns. The future advent of genome-wide approaches in clinical practice could allow the identification of additional driver gene mutations and potential MRD markers suitable for prognosis and innovative therapeutic procedures [46]. NGS offers precise gene sequencing, but what is the true impact of the discovered mutations on leukemogenesis? These questions remain unclear to clinicians and scientists; therefore, one of the imperfections of NGS is the inability to determine the impact of a particular mutation. Hence, the challenge of introducing it into routine clinical practice [35]. Furthermore, a distinction must also be made between leukemia-related somatic mutations and clonal hematopoiesis of indeterminate potential (CHIP). CHIP by definition is a process associated with the aging of hematopoietic cells, in which they form clones that have acquired leukemia-related mutations with an allelic frequency of 2% or more. Thus, in AML patients, even in the period of clinical remission, certain mutations of genes such as TET2, ASXL1, RUNX1, IDH, DNMT3A and others may be present [47]. Along this line, another important aspect to consider when interpreting NGS assays includes germline mutations in certain genes that may be involved in leukemogenesis. The role of these germline mutations is not always clear; thus, their numbers are likely to grow in the future as NGS progresses. For example, one study reported germline p53 mutations in 6 of 107 AML patients after cancer treatment [48]. Thus, the role of this mutation in leukemogenesis is undoubtedly clear. It will be of great importance in the future to create updated and extensive cancer databases that would include all mutations that can initiate the leukemogenic process. Moreover, NGS analysis in AML patients is of particular importance applied to the IDH1, IDH2 or FLT3-ITD/TKD genes, as they may represent a hot spot for target therapy. NGS as a newly introduced method will proceed through many more processes of intensive comparative analysis with existing methods of molecular diagnosis before being applied in clinical practice. A multidisciplinary approach must be taken to overcome technical, economic and organizational aspects.References
- De Kouchkovsky, I.; Abdul-Hay, M. Acute myeloid leukemia: A comprehensive review and 2016 update. Blood Cancer J. 2016, 6, e441.
- Grove, C.S.; Vassiliou, G.S. Acute myeloid leukaemia: A paradigm for the clonal evolution of cancer? Dis. Models Mech. 2014, 7, 941–951.
- Goel, H.; Rahul, E.; Gupta, I.; Chopra, A.; Ranjan, A.; Gupta, A.K.; Meena, J.P.; Viswanathan, G.K.; Bakhshi, S.; Misra, A. Molecular and genomic landscapes in secondary & therapy related acute myeloid leukemia. Am. J. Blood Res. 2021, 11, 472.
- Höllein, A.; Nadarajah, N.; Meggendorfer, M.; Jeromin, S.; Kern, W.; Haferlach, C.; Haferlach, T. Molecular characterization of aml with runx1-runx1t1 at diagnosis and relapse reveals net loss of co-mutations. HemaSphere 2019, 3, e178.
- Liquori, A.; Ibañez, M.; Sargas, C.; Sanz, M.Á.; Barragán, E.; Cervera, J. Acute promyelocytic leukemia: A constellation of molecular events around a single pml-rara fusion gene. Cancers 2020, 12, 624.
- Yang, J.J.; Park, T.S.; Wan, T.S. Recurrent cytogenetic abnormalities in acute myeloid leukemia. Cancer Cytogenet. 2017, 1541, 223–245.
- Cancer Genome Atlas Research Network. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N. Engl. J. Med. 2013, 368, 2059–2074.
- Kansal, R. Toward integrated genomic diagnosis in routine diagnostic pathology by the world health organization classification of acute myeloid leukemia. J. Clin. Haematol. 2020, 1, 2.
- Carter, J.L.; Hege, K.; Yang, J.; Kalpage, H.A.; Su, Y.; Edwards, H.; Hüttemann, M.; Taub, J.W.; Ge, Y. Targeting multiple signaling pathways: The new approach to acute myeloid leukemia therapy. Signal Transduct. Target. Ther. 2020, 5, 288.
- Calabrese, C.; Panuzzo, C.; Stanga, S.; Andreani, G.; Ravera, S.; Maglione, A.; Pironi, L.; Petiti, J.; Shahzad Ali, M.; Scaravaglio, P. Deferasirox-dependent iron chelation enhances mitochondrial dysfunction and restores p53 signaling by stabilization of p53 family members in leukemic cells. Int. J. Mol. Sci. 2020, 21, 7674.
- Panuzzo, C.; Signorino, E.; Calabrese, C.; Ali, M.S.; Petiti, J.; Bracco, E.; Cilloni, D. Landscape of tumor suppressor mutations in acute myeloid leukemia. J. Clin. Med. 2020, 9, 802.
- Saliba, A.N.; John, A.J.; Kaufmann, S.H. Resistance to venetoclax and hypomethylating agents in acute myeloid leukemia. Cancer Drug Resist. 2021, 4, 125.
- Liu, X.; Gong, Y. Isocitrate dehydrogenase inhibitors in acute myeloid leukemia. Biomark. Res. 2019, 7, 22.
- Sekeres, M.A.; Guyatt, G.; Abel, G.; Alibhai, S.; Altman, J.K.; Buckstein, R.; Choe, H.; Desai, P.; Erba, H.; Hourigan, C.S. American society of hematology 2020 guidelines for treating newly diagnosed acute myeloid leukemia in older adults. Blood Adv. 2020, 4, 3528–3549.
- Voso, M.T.; Ottone, T.; Lavorgna, S.; Venditti, A.; Maurillo, L.; Lo-Coco, F.; Buccisano, F. Mrd in aml: The role of new techniques. Front. Oncol. 2019, 9, 655.
- Schuurhuis, G.J.; Heuser, M.; Freeman, S.; Béné, M.-C.; Buccisano, F.; Cloos, J.; Grimwade, D.; Haferlach, T.; Hills, R.K.; Hourigan, C.S. Minimal/measurable residual disease in aml: A consensus document from the european leukemianet mrd working party. Blood J. Am. Soc. Hematol. 2018, 131, 1275–1291.
- Hauwel, M.; Matthes, T. Minimal residual disease monitoring: The new standard for treatment evaluation of haematological malignancies? Swiss Med. Wkly. 2014, 144, w13907.
- Gabert, J.; Beillard, E.; van der Velden, V.H.J.; Bi, W.; Grimwade, D.; Pallisgaard, N.; Barbany, G.; Cazzaniga, G.; Cayuela, J.M.; Cavé, H.; et al. Standardization and quality control studies of ‘real-time’ quantitative reverse transcriptase polymerase chain reaction of fusion gene transcripts for residual disease detection in leukemia—A Europe against cancer program. Leukemia 2003, 17, 2318–2357.
- Aitken, M.J.; Ravandi, F.; Patel, K.P.; Short, N.J. Prognostic and therapeutic implications of measurable residual disease in acute myeloid leukemia. J. Hematol. Oncol. 2021, 14, 137.
- Ossenkoppele, G.; Schuurhuis, G.J. MRD in AML: Does it already guide therapy decision-making? Hematol. 2014 Am. Soc. Hematol. Educ. Program Book 2016, 2016, 356–365.
- Döhner, H.; Estey, E.; Grimwade, D.; Amadori, S.; Appelbaum, F.R.; Büchner, T.; Dombret, H.; Ebert, B.L.; Fenaux, P.; Larson, R.A. Diagnosis and management of aml in adults: 2017 eln recommendations from an international expert panel. Blood J. Am. Soc. Hematol. 2017, 129, 424–447.
- Chendamarai, E.; Balasubramanian, P.; George, B.; Viswabandya, A.; Abraham, A.; Ahmed, R.; Alex, A.A.; Ganesan, S.; Lakshmi, K.M.; Sitaram, U. Role of minimal residual disease monitoring in acute promyelocytic leukemia treated with arsenic trioxide in frontline therapy. Blood J. Am. Soc. Hematol. 2012, 119, 3413–3419.
- Chen, Z.; Tong, Y.; Li, Y.; Gao, Q.; Wang, Q.; Fu, C.; Xia, Z. Development and validation of a 3-plex rt-qpcr assay for the simultaneous detection and quantitation of the three pml-rara fusion transcripts in acute promyelocytic leukemia. PLoS ONE 2015, 10, e0122530.
- Willekens, C.; Blanchet, O.; Renneville, A.; Cornillet-Lefebvre, P.; Pautas, C.; Guieze, R.; Ifrah, N.; Dombret, H.; Jourdan, E.; Preudhomme, C. Prospective long-term minimal residual disease monitoring using rq-pcr in runx1-runx1t1-positive acute myeloid leukemia: Results of the french cbf-2006 trial. Haematologica 2016, 101, 328.
- Jourdan, E.; Boissel, N.; Chevret, S.; Delabesse, E.; Renneville, A.; Cornillet, P.; Blanchet, O.; Cayuela, J.-M.; Recher, C.; Raffoux, E. Prospective evaluation of gene mutations and minimal residual disease in patients with core binding factor acute myeloid leukemia. Blood J. Am. Soc. Hematol. 2013, 121, 2213–2223.
- Puckrin, R.; Atenafu, E.G.; Claudio, J.O.; Chan, S.; Gupta, V.; Maze, D.; McNamara, C.; Murphy, T.; Schuh, A.C.; Yee, K. Measurable residual disease monitoring provides insufficient lead-time to prevent morphological relapse in the majority of patients with core-binding factor acute myeloid leukemia. Haematologica 2021, 106, 56–63.
- Falini, B.; Brunetti, L.; Sportoletti, P.; Martelli, M.P. Npm1-mutated acute myeloid leukemia: From bench to bedside. Blood 2020, 136, 1707–1721.
- Gorello, P.; Cazzaniga, G.; Alberti, F.; Dell’Oro, M.; Gottardi, E.; Specchia, G.; Roti, G.; Rosati, R.; Martelli, M.; Diverio, D. Quantitative assessment of minimal residual disease in acute myeloid leukemia carrying nucleophosmin (npm1) gene mutations. Leukemia 2006, 20, 1103–1108.
- Forghieri, F.; Comoli, P.; Marasca, R.; Potenza, L.; Luppi, M. Minimal/measurable residual disease monitoring in npm1-mutated acute myeloid leukemia: A clinical viewpoint and perspectives. Int. J. Mol. Sci. 2018, 19, 3492.
- Tiong, S.; Dillon, R.; Ivey, A.; Kok, C.H.; Kuzich, J.A.; Thiagarajah, N.; Bajel, A.; Potter, N.; Smith, M.; Hemmaway, C. The natural history of npm1mut measurable residual disease (MRD) positivity after completion of chemotherapy in acute myeloid leukemia (AML). Blood 2020, 136, 25–27.
- Lussana, F.; Caprioli, C.; Stefanoni, P.; Pavoni, C.; Spinelli, O.; Buklijas, K.; Michelato, A.; Borleri, G.; Algarotti, A.; Micò, C. Molecular detection of minimal residual disease before allogeneic stem cell transplantation predicts a high incidence of early relapse in adult patients with npm1 positive acute myeloid leukemia. Cancers 2019, 11, 1455.
- Balsat, M.; Renneville, A.; Thomas, X.; de Botton, S.; Caillot, D.; Marceau, A.; Lemasle, E.; Marolleau, J.-P.; Nibourel, O.; Berthon, C. Postinduction minimal residual disease predicts outcome and benefit from allogeneic stem cell transplantation in acute myeloid leukemia with npm1 mutation: A study by the acute leukemia french association group. J. Clin. Oncol. 2017, 35, 185–193.
- Behjati, S.; Tarpey, P.S. What is next generation sequencing? Arch. Dis. Child. Educ. Pract. 2013, 98, 236–238.
- Anderson, M.W.; Schrijver, I. Next generation DNA sequencing and the future of genomic medicine. Genes 2010, 1, 38–69.
- Bacher, U.; Shumilov, E.; Flach, J.; Porret, N.; Joncourt, R.; Wiedemann, G.; Fiedler, M.; Novak, U.; Amstutz, U.; Pabst, T. Challenges in the introduction of next-generation sequencing (ngs) for diagnostics of myeloid malignancies into clinical routine use. Blood Cancer J. 2018, 8, 113.
- Vainchenker, W.; Kralovics, R. Genetic basis and molecular pathophysiology of classical myeloproliferative neoplasms. Blood J. Am. Soc. Hematol. 2017, 129, 667–679.
- Vannucchi, A.M.; Lasho, T.; Guglielmelli, P.; Biamonte, F.; Pardanani, A.; Pereira, A.; Finke, C.; Score, J.; Gangat, N.; Mannarelli, C. Mutations and prognosis in primary myelofibrosis. Leukemia 2013, 27, 1861–1869.
- Tefferi, A.; Lasho, T.L.; Guglielmelli, P.; Finke, C.M.; Rotunno, G.; Elala, Y.; Pacilli, A.; Hanson, C.A.; Pancrazzi, A.; Ketterling, R.P. Targeted deep sequencing in polycythemia vera and essential thrombocythemia. Blood Adv. 2016, 1, 21–30.
- Papaemmanuil, E.; Gerstung, M.; Bullinger, L.; Gaidzik, V.I.; Paschka, P.; Roberts, N.D.; Potter, N.E.; Heuser, M.; Thol, F.; Bolli, N. Genomic classification and prognosis in acute myeloid leukemia. N. Engl. J. Med. 2016, 374, 2209–2221.
- Thol, F.; Kölking, B.; Damm, F.; Reinhardt, K.; Klusmann, J.H.; Reinhardt, D.; von Neuhoff, N.; Brugman, M.H.; Schlegelberger, B.; Suerbaum, S. Next-generation sequencing for minimal residual disease monitoring in acute myeloid leukemia patients with flt3-itd or npm1 mutations. Genes Chromosomes Cancer 2012, 51, 689–695.
- Morita, K.; Kantarjian, H.M.; Wang, F.; Yan, Y.; Bueso-Ramos, C.; Sasaki, K.; Issa, G.C.; Wang, S.; Jorgensen, J.; Song, X. Clearance of somatic mutations at remission and the risk of relapse in acute myeloid leukemia. J. Clin. Oncol. 2018, 36, 1788.
- Kohlmann, A.; Nadarajah, N.; Alpermann, T.; Grossmann, V.; Schindela, S.; Dicker, F.; Roller, A.; Kern, W.; Haferlach, C.; Schnittger, S. Monitoring of residual disease by next-generation deep-sequencing of runx1 mutations can identify acute myeloid leukemia patients with resistant disease. Leukemia 2014, 28, 129–137.
- Jongen-Lavrencic, M.; Grob, T.; Hanekamp, D.; Kavelaars, F.G.; Al Hinai, A.; Zeilemaker, A.; Erpelinck-Verschueren, C.A.; Gradowska, P.L.; Meijer, R.; Cloos, J. Molecular minimal residual disease in acute myeloid leukemia. N. Engl. J. Med. 2018, 378, 1189–1199.
- Mertens, F.; Johansson, B.; Fioretos, T.; Mitelman, F. The emerging complexity of gene fusions in cancer. Nat. Rev. Cancer 2015, 15, 371–381.
- Leisch, M.; Jansko, B.; Zaborsky, N.; Greil, R.; Pleyer, L. Next generation sequencing in aml—On the way to becoming a new standard for treatment initiation and/or modulation? Cancers 2019, 11, 252.
- Metzeler, K.H.; Herold, T.; Rothenberg-Thurley, M.; Amler, S.; Sauerland, M.C.; Görlich, D.; Schneider, S.; Konstandin, N.P.; Dufour, A.; Bräundl, K. Spectrum and prognostic relevance of driver gene mutations in acute myeloid leukemia. Blood J. Am. Soc. Hematol. 2016, 128, 686–698.
- Bhatnagar, B.; Eisfeld, A.K.; Nicolet, D.; Mrózek, K.; Blachly, J.S.; Orwick, S.; Lucas, D.M.; Kohlschmidt, J.; Blum, W.; Kolitz, J.E. Persistence of dnmt 3a r882 mutations during remission does not adversely affect outcomes of patients with acute myeloid leukaemia. Br. J. Haematol. 2016, 175, 226–236.
- Zebisch, A.; Lal, R.; Müller, M.; Lind, K.; Kashofer, K.; Girschikofsky, M.; Fuchs, D.; Wölfler, A.; Geigl, J.B.; Sill, H. Acute myeloid leukemia with tp53 germ line mutations. Blood J. Am. Soc. Hematol. 2016, 128, 2270–2272.