Mitochondrial proteins are encoded by both nuclear and mitochondrial DNA. While some of the essential subunits of the oxidative phosphorylation (OXPHOS) complexes responsible for cellular ATP production are synthesized directly in the mitochondria, most mitochondrial proteins are first translated in the cytosol and then imported into the organelle using a sophisticated transport system. These proteins are directed mainly by targeting presequences at their N-termini. These presequences need to be cleaved to allow the proper folding and assembly of the pre-proteins into functional protein complexes. In the mitochondria, the presequences are removed by several processing peptidases, including the mitochondrial processing peptidase (MPP), the inner membrane processing peptidase (IMP), the inter-membrane processing peptidase (MIP), and the mitochondrial rhomboid protease (Pcp1/PARL). Their proper functioning is essential for mitochondrial homeostasis as the disruption of any of them is lethal in yeast and severely impacts the lifespan and survival in humans.
- mitochondrial processing peptidases
- MPP
- MIP
- IMP
- mitochondrial rhomboid protease
- mitochondrial disease
1. Introduction
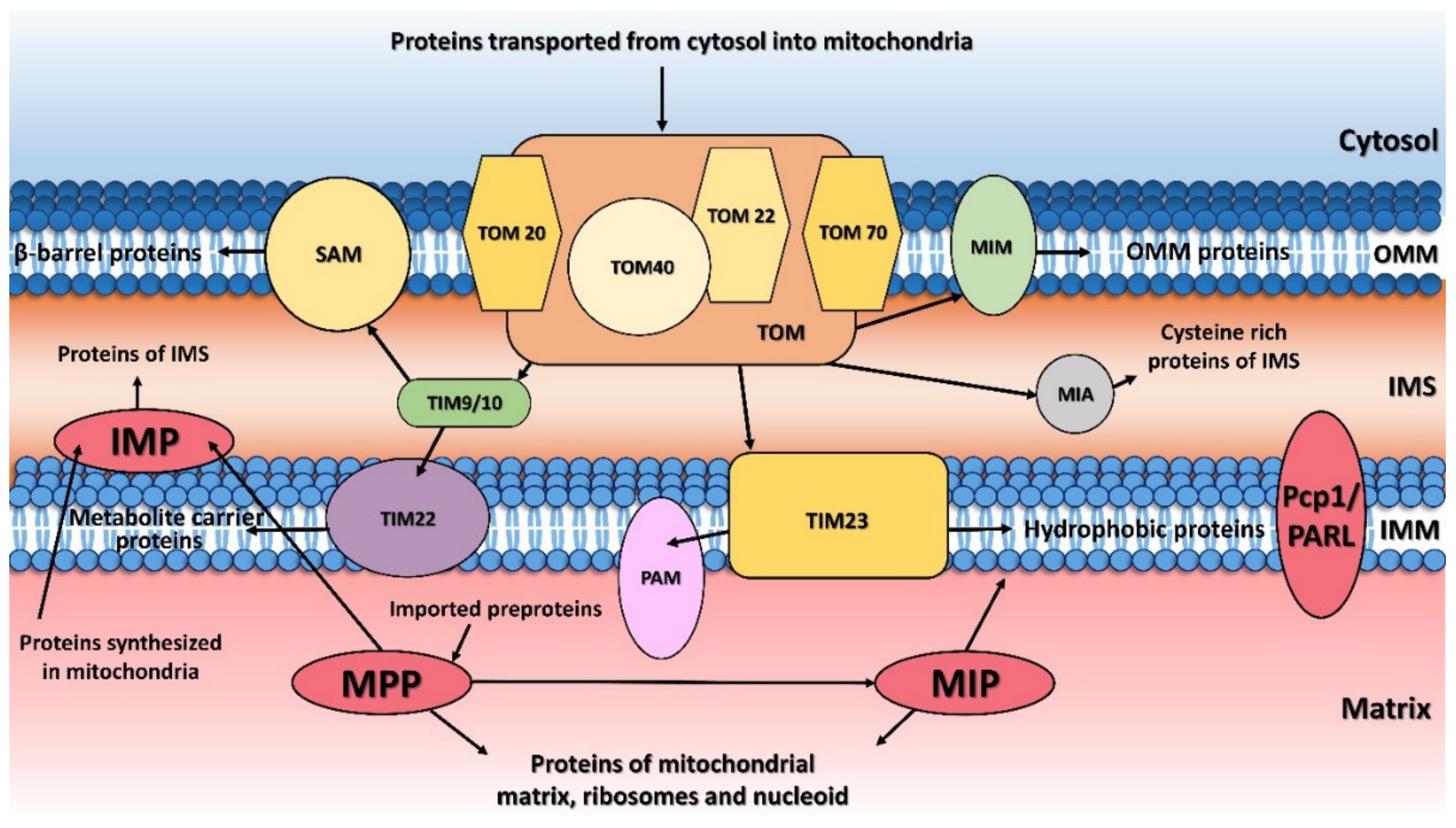
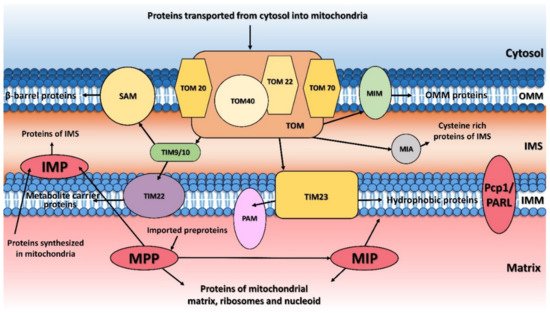
2. Mitochondrial Processing Peptidase
The principal responsibility of the mitochondrial processing peptidase is to remove the N-terminal targeting presequences of proteins imported into the mitochondria (Figure 1). Although the presequences can vary in length and amino-acid composition, they have several common properties. They are all predicted to form an amphiphilic α-helix [20][21][21,22], have an overall positive charge, and have an arginine residue at position -2 or -3 from the cleavage site [22][23].
MPP is a hetero-dimeric protein consisting of two subunits, α and β, which are referred to as PMPCA and PMPCB in humans [11][12][7,8]. These subunits together create a large substrate-binding cavity with a Zn2+-binding site on the MPPβ subunit. The site itself is created by a conserved HxxEHx76E motif in the MPPβ subunit; the mutation of any of these residues eliminates Zn2+ binding and blocks the peptidase activity. Although the β subunit contains the entirety of the catalytic site, the cooperation of action of both MPP subunits is required for proper processing of pre-proteins. The most conserved part of all known MPPα subunits is a glycine-rich loop (GRL; residues G284GGGSFSAGGPGKGMYS300 in yeast MPPα), which is essential for substrate binding [23][24][17,18] and which moves the precursor protein towards the active site through a multistep process [25][19]. An electrostatic analysis of MPP complexed to a peptide substrate showed that the binding cavity was strongly negatively-charged while the substrate peptide is positively charged.
Deletion of both MPP encoding genes (MPPA and MPPB) is incompatible with the viability of S. cerevisiae under any and all growth conditions, including even anaerobic growth [26][27][24,25]. In humans, mutations in either PMPCA or PMPCB cause mitochondrial diseases that are characterized by neurological disorder with an early childhood onset and a severe neurodegenerative course [28][29][30][26-28] (Table 1).
| Processing Peptidase | Protein Variant | Disease, Symptoms | Ref. |
|---|---|---|---|
| PMPCA | Homozygous mutation: c.1129G>A (p.Ala377Thr) Heterozygous mutations: c.287C>T (p.Ser96Leu) with c.1543G>A (p.Gly515Arg) |
SCAR2 with non- or slowly progressive cerebellar ataxia and developmental delay | [31][38] |
| Homozygous mutation: c.766G>A (p.Val256Met) | slowly progressive SCAR2 without intellectual disability | [32][47] | |
| Heterozygous mutation: c.677C>T (p.Arg223Cys) with c.853del (p.Asp285Ilefs*16) | SCAR2 with progressive cerebellar ataxia and onset in infancy | [18] | |
| Heterozygous mutations: c.1066G>A (p.Gly356Ser) with c.1129G>A (p.Ala377Thr) | SCAR2 with progressive, extensive brain atrophy, muscle weakness, visual impairment, respiratory defects | [33][48] | |
| Homozygous mutation: c.553C>T (p.Arg185Thr) | SCAR2 with psychomotor delay | [34][46] | |
| PMPCB | Heterozygous mutations: c.523C>T (p.Arg175Cys) with c.601G>C (p.Ala201Pro); c.524G>A (p.Arg175His) with c.530T>G (p.Val177Gly) Homozygous mutation: c.1265T>C (p.Ile422Thr) |
Prominent cerebellar atrophy in early childhood | [35][50] |
| IMMP2L | Duplication: 46,XY,dup(7)(q22.1-q31.1) | GTS/TS | [15] |
| Deletions ranged from ~49 kb to ~337 kb | Neurological disorders (ADHD, GTS/TS, OCD, ASD, Asperger′s syndrome, schizophrenia and developmental delay) | [36][37][38][39][53,54,55,56] | |
| Base pair change | Autism | [40][57] | |
| Copy number variation | Alzheimer′s disease | [41][58] | |
| Downregulation | Prostate cancer | [42][59] | |
| MIP | Homozygous SNV: p.K343E Heterozygous SNVs: p.L582R with p.L71Q; p.E602* with p.L306 and p.H512D with 1.4-Mb deletion of 13q12.12 |
LVNC and developmental delay, seizures, hypotonia | [43][60] |
| Heterozygous mutation: c.916C > T (p.Leu306Phe) with c.1970 + 2 T>A (p.Ala658Lysfs*38) | Developmental delay, hypotonia and intellectual disability | [44][61] | |
| Hypomethylation | Metabolic syndrome | [45][62] | |
| Downregulation | Prostate cancer | [42][59] | |
| PARL | Reduced levels | Type 2 diabetes | [19] |
| Leu262Val polymorphism | Increased plasma insulin concentration | [46][63] | |
| Mutation: c.230G>A (p.Ser77Asn) | Parkinson′s disease | [47][64] |
3. Mitochondrial Inner Membrane Peptidase
The mitochondrial inner membrane peptidase is responsible for the maturation of proteins transported into the mitochondrial inter-membrane space (Figure 1) [48][49][50][51][65–68]. These include mature proteins synthesized both within the mitochondria (e.g., yeast mitochondrially encoded subunit 2 of cytochrome c oxidase, Cox2), or nuclear-encoded proteins synthesized in the cytosol and then transported into the mitochondria (e.g., yeast cytochrome b2, Cyb2, cytochrome c1, Cyt1, and NADH cytochrome b5 reductase, Mcr1).
Structurally, IMP consists of two subunits; in humans, these are IMMP1L (inner membrane mitochondrial peptidase 1-like) and IMMP2L (inner membrane mitochondrial peptidase 2-like) [49][66], and in S. cerevisiae there are three subunits, Imp1, Imp2, and Som1 [50][67]. Although the sequence identities between the individual yeast and human IMP homologues are relatively low (between 25-37%), their tertiary structures share a number of common features. All four IMP homologues are predicted to have a membrane-anchored α-helical N-terminal domain and a catalytic C-terminal domain. The yeast Imp1 and Imp2 subunits share 31% amino-acid sequence identity and both possess catalytic activity and are bound to the inner mitochondrial membrane [52][53][54][70–72]. The catalytic domain possesses a catalytic Ser/Lys dyad, which is present in all four proteins and is structurally located in the C-terminal region [51][55][68,73]. The third yeast subunit, Som1, most likely serves to recognize substrates and was shown to physically interact with Imp1 [51][56][68,74]. Surprisingly, Som1 seems to be important for the Imp1-mediated proteolytic processing of Cox2 and Mcr1, but not for the maturation of the Cyb2 and Cyt1 cytochromes processed by the Imp2 subunit [51][56][68,74].
The currently known natural Imp1 substrates all possess a characteristic [I/V][H/D/F/M][N](↓)[D/E] amino-acid motif surrounding the cleavage site (indicated by ↓) [67]. Although the substrate specificities for Imp1 and Imp2 do not overlap, there are recognizable similarities between the protein precursors that they cleave. These include a hydrophobic residue at position -3 from the cleavage site and, for the nucleus-encoded substrates, the distances between the transmembrane segment and the cleavage site are also preserved. The accessibility of the cleavage site to the peptidase is also a prerequisite for cleavage by IMP [50][67].
In humans, the IMP homolog, IMMP2L has a 41% similarity to the yeast Imp2 subunit and a 90% similarity to the mouse IMMP2L [49][66]. It is composed of 175 amino acids with a gene of 860 kb located on chromosome 7q (AUTS1 locus), whose integrity has been shown to be critical for the development of autism spectrum disorders (ASDs). IMMP2L is expressed at a basal level in all human tissues except for the lungs and liver of adults [15][36][15,53]. Mutations associated with the gene encoding IMMP2L have been observed in several neurodegenerative diseases, including Gills de la Tourette syndrome or Tourette′s syndrome (GTS/TS), attention-deficit hyperactivity disorder (ADHD), ASD, and schizophrenia [36][37][39][57][58][53,54,56,75,76] (see Table 1 above).
4. Mitochondrial Intermediate Peptidase
The mitochondrial intermediate peptidase is important for the maturation of a subgroup of precursor proteins imported into the mitochondrial matrix or embedded into the mitochondrial inner membrane [59][43]. These pre-proteins are first processed by MPP and only afterwards by MIP, which cleaves an additional octapeptide following MPP cleavage. The cleavage site targeted by MIP is characterized by an RX(↓)(F/L/I)XX(T/S/G)XXXX(↓) motif [48][65] and is located at the C-terminus of a leader peptide (↓). Active MIP is a soluble monomer of 75 kDa in yeast and 81 kDa in humans. Its proteolytic activity is stimulated by manganese, magnesium and calcium ions while 1 mM Co2+, Fe2+ or Zn2+ completely inhibits it. Unlike MPP, MIP is also sensitive to N-ethylmaleimide (NEM) and other sulfhydryl reagents [60][88].
Positioning at the substrate N-terminus and a large hydrophobic residue (phenylalanine, leucine and isoleucine) at position -8 from the cleavage site are both essential features for cleavage by MIP; this type of substrate specificity is not shared by any other known peptidase [48][65].
In S. cerevisiae, mitochondrial oxidative phosphorylation is severely affected when mip1 is missing. Branda et al. [48][65] showed that at least three vital components of the yeast mitochondrial gene expression machinery—mitochondrial small ribosomal subunit protein MrpS28, single-stranded DNA-binding protein Rim1, and elongation factor Tuf1—are processed by MIP. These proteins are essential for maintaining mitochondrial protein synthesis and mitochondrial DNA replication, which explains why the loss of mip1 impairs the mitochondrially encoded OXPHOS subunits. MIP1 disruption also results in the failure of at least two yeast nuclear-encoded respiratory chain components, the cytochrome c oxidase subunit 4 (Cox4) and the Rieske iron-sulfur protein of cytochrome bc1 catalytic subunit, to be cleaved [59][43].
In humans, MIP is encoded by the MIPEP gene, which contains 19 exons and is located on chromosome 13q12.12 [61][89]. MIPEP is expressed at high levels in energy-dependent tissues, such as the heart, brain, skeletal muscles, and pancreas [61][62][63][89–91]. Previously, some patients were reported with mutations in MIPEP which may have been linked to their diagnoses, but the first study showing that MIPEP is truly involved in a human disease was published in 2016 by Eldomery et al. [43][60] (see Table 1 above). They identified several single nucleotide variants (SNVs) in the MIPEP gene that caused a loss of MIP function in four unrelated patients suffering from an oxidative phosphorylation deficiency. Further studies in human fibroblasts showed that MIP has an important role in OXPHOS function since its loss impaired the processing of several OXPHOS subunits, including the OXPHOS complexes I, IV and V [44][61].