Lung cancer (LC) is one of the leading causes of cancer occurrence and mortality worldwide. Treatment of patients with advanced and metastatic LC presents a significant challenge, as malignant cells use different mechanisms to resist chemotherapy. Drug resistance (DR) is a complex process that occurs due to a variety of genetic and acquired factors. Identifying the mechanisms underlying DR in LC patients and possible therapeutic alternatives for more efficient therapy is a central goal of LC research. Advances in nanotechnology resulted in the development of targeted and multifunctional nanoscale drug constructs. The possible modulation of the components of nanomedicine, their surface functionalization, and the encapsulation of various active therapeutics provide promising tools to bypass crucial biological barriers. These attributes enhance the delivery of multiple therapeutic agents directly to the tumor microenvironment (TME), resulting in reversal of LC resistance to anticancer treatment.
- nanomedicine
- drug resistance
- lung cancer
1. Introduction
The therapeutic effectiveness of chemotherapeutic agents is limited due to the development of DR in cancer cells [5][1]. Cancer DR is the ability of the tumor cells to develop a certain mechanism to overcome and resist the cytotoxic or inhibitory effect of the chemotherapeutic agent and therefore reduce the effectiveness of chemotherapy [6][2]. Currently, the failure of chemotherapy due to DR accounts for 90% of clinical metastasis cases [6][2]. To overcome DR, chemotherapeutic agents need to be administered at larger doses with higher frequency, which in turn may result in increased toxicity and lower patient survival rate. Alternatively, a combination of two or more chemotherapeutic agents may be administered to achieve a synergistic effect and reduce the rate of DR [7][3]. This approach has improved the effectiveness of chemotherapy but has not yet eliminated the side effects associated with non-specific uptake by normal cells.
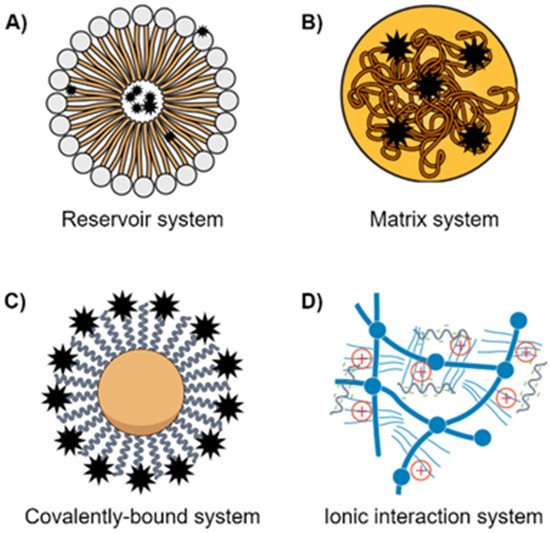
2. Nanomedicine Applications in Management of Lung Cancer Drug Resistance
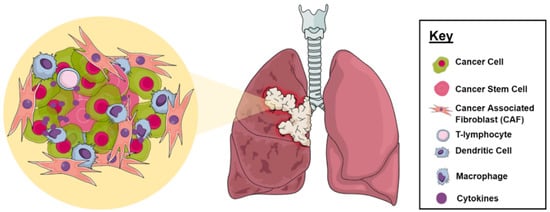
2.1. Tumor Microenvironment
2.2. Multidrug Resistance
Tumor multidrug resistance (MDR) remains a major obstacle that continues to hinder the effective progression of current curative cancer therapy in LC [42][35]. Innate and acquired phenotypes have been frequently identified as major cancer cell defense mechanisms following exposure to chemotherapeutic regimens [43][36]. Until now, several MDR mechanisms have been increasingly linked to members of the ATP-binding cassette (ABC) membrane pumps with 48 identified genes [44][37]. A number of these efflux transporters, including P-glycoprotein (P-gp; ABCB1; MDR1), breast cancer resistance protein (BCRP; ABCG2), and MDR-associated protein 1 (MRP1; ABCC1), have been recognized as reducing the efficacy of anticancer agents in tumor cells through a noticeable decrease in their intracellular accumulation in an ATP-dependent manner (Figure 3) [45][38]. Commonly used chemotherapeutic agents, including taxanes, platinum compounds, and gemcitabine, fall victim to these pathways [46,47,48,49][39][40][41][42].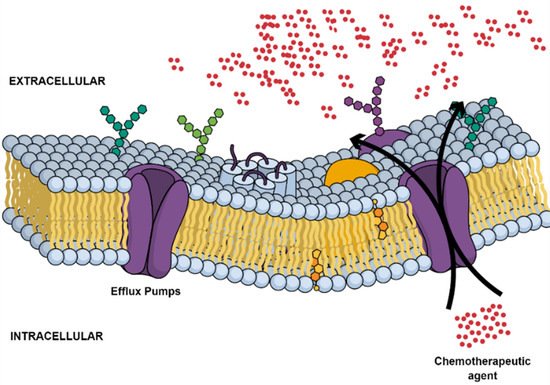
2.3. Cancer Stem Cells
2.4. Metabolic Inactivation of the Anticancer Drugs
Drug detoxification is considered a key resistance mechanism in several types of malignant tumors (Figure 4). Each population of cancer cells can respond differently to anticancer drugs due to the associated genomic variation [78][60]. The metabolism of chemotherapeutic agents can progress intracellularly and/or extracellularly, eventually affecting the overall efficacy of the given anticancer agent [79][61].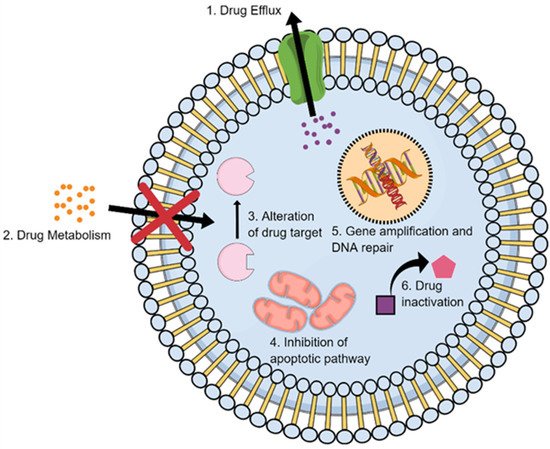
2.5. Inhibition of the Cell Death
2.6. Alteration of Drug Targets
Resistance to chemotherapeutic agents can be due to alteration in their targets at the tumor sites. These changes occur due to molecular modifications that may begin by mutation in DNA and alterations in protein expression, resulting in a decrease in the affinity of the drugs with their binding targets and DR (Figure 4). For example, treatment of SCLC with DOX in combination with platinum drugs inhibits the topoisomerase enzymes in the cells by intercalation between the DNA bases, causing inhibition to the enzyme gyrase that is responsible unwinding the structure of DNA during the DNA replication and ultimately causing DNA breakage. Many of resistant cancer cells can survive this treatment by modifying topoisomerase II gene expression and hence altering the target of DOX [78,139][60][81]. A similar DR mechanism was also reported for anticancer drugs that target specific signaling kinases, such as the epidermal growth factor receptor (EGFR) family [26,140,141][22][82][83]. In this case, a mutation commonly occurs in the receptor kinase, leading to over-activation of these kinases and their downstream signaling molecules such as Ras, Src, and MEK. Many of these kinases become constitutively active and promote uncontrollable cell growth. In some cancers, if the drug targets molecules of the signaling pathways, the resistant cancer cells tend to activate alternative molecules. The mutations in the EGFR in anaplastic lymphoma kinase (ALK) fusion gene-positive LC after the patient was treated with crizotinib serve as an example. Acquired resistance to the drug occurred via (ALK)-mutations, such as EGFR (L1196M and C1156Y), and some patients had other mechanisms of resistance with both mutations and increase in ALK gene copy number [142,143,144][84][85][86]. The single-nucleotide mutations, such as L1196 and G1269A, were reported in some cases to cause crizotinib resistance in NSCLC [145][87]. However, sometimes, the same effect of the mutation that causes over-activation can be found via gene overexpression. Overexpression of certain receptors in some LCs with a mutation in the EGFR tyrosine kinase domain causes drug-acquired resistance that may occur after the long-term use of drugs inhibitors targeting this kinase [145][87]. EGFR-targeted liposomal nanoparticles (EGFR-LP) were developed for the treatment of NSCLC resistance to drugs as erlotinib and afatinib, determined by mutations in the tyrosine kinase (TK) domain of EGFR [146][88]. Ramanathan and colleagues have re-ported a novel DNA-based colorimetric assay for the detection of early EGFR mutation using unmodified gold nanoparticles (GNPs) [147][89].2.7. Enhancing DNA Repair
DNA repair involves a tangled network of repair mechanisms dictated by the specific kind of stimuli and damage to which cells are exposed (Figure 4). These mechanisms include mismatch repair (MMR), nucleotide excision repair (NER), base excision repair (BER), direct reversal (MGMT, ABH2, ABH3), homologous recombination (HR) and nonhomologous end joining (NHEJ) pathways. For instance, ionizing radiation induces double-strand breaks (DSBs) mainly repaired by nonhomologous end joining (NHEJ) pathways. On the other hand, mono- and bifunctional alkylators can induce DNA-base modifications interfering with DNA synthesis, which can be reversed in a mismatched repair-dependent manner [44,164,165][37][90][91]. Inhibition of DNA repair systems may be a potential strategy to sensitize cancer cells to chemotherapeutic drugs and increase their efficacy. However, even if disrupting DNA repair systems may block the resistance to chemotherapeutic agents, it can also be responsible for the development of new mutations due to genomic instability [166][92]. CIS-resistant cancer cells showed higher levels of DNA damage repair. In addition, it was noted that inhibition of NER pathways can significantly enhance tumor cells’ sensitivity to CIS. The enhanced DNA repair capability in lung-CSCs was associated with an extensive activation of DNA repair genes in response to CIS treatment, suggesting it may be the main mechanism involved in resistance insurgence [167,168][93][94]. Studies have also highlighted an inverse correlation of ERCC1 (NER pathways) with response to platinum therapy in LC [169][95]. Apurinic/apyrimidinic endonuclease 1 (APE1) is considered a crucial BER pathway protein due to its activity as intermediate in the processing of potentially cytotoxic DNA damage sites. Moreover, APE1 seems to have a dual role, depending on its cellular localization, where it carries out DNA repair in the nucleus. However, in the cytoplasm, its primary role is assumed to be the regulation of mitochondrial DNA repair, possibly together with the regulation of various transcription factors. In LC cells, APE1 is often overexpressed, especially in CIS-resistant cancers [170,171][96][97].2.8. Gene Amplification
2.9. Epigenetic Alteration Caused Drug Resistance
Although all cells of the human body have the same exact genes, epigenetic alterations regulate the way genome can be read. These are changes in the chemical structure of DNA that do not change the nucleotide coding sequence but have a profound effect on gene expression. Epigenetic alterations may occur due to the adding of and exposure to environmental factors, such as diet, exercise, drugs, and chemicals [187,188,189][109][110][111]. Methylation and acetylation of DNA are two well-studied epigenetic events that significantly alter the expression of genes, resulting in the upregulation of oncogenes and/or downregulation of tumor suppressor genes and development of cancer DR [190][112]. In eukaryotes, histones mainly serve as a structure guide for several enzymes to provide the necessary platform for RNA polymerase access to its target. Histone acetyltransferases (HATs) and histone deacetylases (HDACs) are essential enzymes that regulate histone acetylation, which is the pivotal focus of several studies on post-translational modification mechanisms. Most of the common features displayed by cancerous cells, such as the evasion of apoptosis, increased angiogenesis, and metastasis progression can be linked to epigenetic modulation and to HDAC. A number of studies highlighted the multiple roles of HDAC, suggesting it as a potential target for chemotherapy and establishing the basis for the development and use of HDAC inhibitors (HDACi) as co-adjuvant for many anticancer agents for treatment of NSCLC [191,192][113][114]. PTX co-administration with HDACi SNOH-3 showed reversed DR in PTX-resistant NSCLC cells characterized by overexpression of HDAC1 [193][115]. Sharma et al. demonstrated the ability of a subset of stem-like cells in NSCLC cell lines to undergo chromatin remodeling following treatment with erlotinib and CIS, which allow the development of drug insensitivity [194][116]. However, despite the myriad of pre-clinical work supporting HDACi efficacy as adjuvant of chemotherapy in treatment of NSCLC, they have demonstrated modest efficacy as single agents in clinical trials. The use of nanocarriers for the delivery of epigenetic agents has noticeably enhanced their ability as co-adjuvants to re-sensitize cancer cells after the onset of anticancer DR. Studies on using HDACi-loaded NPs in combination with chemotherapy and radiotherapy demonstrated the enhancement of anti-proliferative effects [195][117]. For example, to improve the bioavailability of the histone deacetylase inhibitor vorinostat (VOR) and its efficacy in the treatment of multidrug resistant cancers, solid lipid NPs (SLNs) were used as carriers. Treatment of resistant LC cell line with VOR-SLNs resulted in improved efficacy, elevated payload capacity, and a sustained release profile. The results also showed that lower doses of VOR-SLNs were required to obtain the same cytotoxic effect as free-VOR [196][118].2.10. Clinical Studies Using Nanotechnology for Management of DR in LC
In 2012, the FDA approved the first nano-formulation for treatment of NSCLC patients, Abraxane, which consists of solvent-free albumin-bound PTX-NPs based on its significant improved clinical trial outcomes [212][119]. Other nano-formulations have been the subject of various clinical trials and showed promising therapeutic outcomes in the treatment of resistant LC (http://www.clinicaltrials.gov).
References
- Jabir, N.R.; Tabrez, S.; Ashraf, G.M.; Shakil, S.; Damanhouri, G.A.; Kamal, M.A. Nanotechnology-based approaches in anticancer research. Int. J. Nanomed. 2012, 7, 4391–4408.
- Mansoori, B.; Mohammadi, A.; Davudian, S.; Shirjang, S.; Baradaran, B. The Different Mechanisms of Cancer Drug Resistance: A Brief Review. Adv. Pharm. Bull. 2017, 7, 339–348.
- Sarkar, S.; Horn, G.; Moulton, K.; Oza, A.; Byler, S.; Kokolus, S.; Longacre, M. Cancer Development, Progression, and Therapy: An Epigenetic Overview. Int. J. Mol. Sci. 2013, 14, 21087–21113.
- Patra, J.K.; Das, G.; Fraceto, L.F.; Campos, E.V.R.; del Pilar Rodriguez-Torres, M.; Acosta-Torres, L.S.; Diaz-Torres, L.A.; Grillo, R.; Swamy, M.K.; Sharma, S.; et al. Nano based drug delivery systems: Recent developments and future prospects. J. Nanobiotechnol. 2018, 16, 71.
- Palazzolo, S.; Bayda, S.; Hadla, M.; Caligiuri, I.; Corona, G.; Toffoli, G.; Rizzolio, F. The Clinical Translation of Organic Nanomaterials for Cancer Therapy: A Focus on Polymeric Nanoparticles, Micelles, Liposomes and Exosomes. Curr. Med. Chem. 2018, 25, 4224–4268.
- Bahman, F.; Pittalà, V.; Haider, M.; Greish, K. Enhanced Anticancer Activity of Nanoformulation of Dasatinib against Triple-Negative Breast Cancer. J. Pers. Med. 2021, 11, 559.
- Awad, N.S.; Haider, M.; Paul, V.; AlSawaftah, N.M.; Jagal, J.; Pasricha, R.; Husseini, G.A. Ultrasound-Triggered Liposomes Encapsulating Quantum Dots as Safe Fluorescent Markers for Colorectal Cancer. Pharmaceutics 2021, 13, 2073.
- Haider, M.; Zaki, K.Z.; El Hamshary, M.R.; Hussain, Z.; Orive, G.; Ibrahim, H.O. Polymeric nanocarriers: A promising tool for early diagnosis and efficient treatment of colorectal cancer. J. Adv. Res. 2021.
- Ahmed, I.S.; El Hosary, R.; Hassan, M.A.; Haider, M.; Abd-Rabo, M.M. Efficacy and Safety Profiles of Oral Atorvastatin-Loaded Nanoparticles: Effect of Size Modulation on Biodistribution. Mol. Pharm. 2018, 15, 247–255.
- Haider, M.; Elsherbeny, A.; Jagal, J.; Hubatová-Vacková, A.; Ahmed, I.S. Optimization and Evaluation of Poly(lactide-co-glycolide) Nanoparticles for Enhanced Cellular Uptake and Efficacy of Paclitaxel in the Treatment of Head and Neck Cancer. Pharmaceutics 2020, 12, 828.
- Kim, E.-S.; Ahn, E.H.; Chung, E.; Kim, D.-H. Recent advances in nanobiotechnology and high-throughput molecular techniques for systems biomedicine. Mol. Cells 2013, 36, 477–484.
- Haider, M.; Abdin, S.M.; Kamal, L.; Orive, G. Nanostructured Lipid Carriers for Delivery of Chemotherapeutics: A Review. Pharmaceutics 2020, 12, 288.
- Blanco, E.; Shen, H.; Ferrari, M. Principles of nanoparticle design for overcoming biological barriers to drug delivery. Nat. Biotechnol. 2015, 33, 941–951.
- Kalyane, D.; Raval, N.; Maheshwari, R.; Tambe, V.; Kalia, K.; Tekade, R.K. Employment of enhanced permeability and re-tention effect (EPR): Nanoparticle-based precision tools for targeting of therapeutic and diagnostic agent in cancer. Mater. Sci. Eng. C 2019, 98, 1252–1276.
- Wang, N.; Cheng, X.; Li, N.; Wang, H.; Chen, H. Nanocarriers and Their Loading Strategies. Adv. Health Mater. 2019, 8, e1801002.
- Rad, H.S.; Monkman, J.; Warkiani, M.E.; Ladwa, R.; O’Byrne, K.; Rezaei, N.; Kulasinghe, A. Understanding the tumor microenvironment for effective immunotherapy. Med. Res. Rev. 2021, 41, 1474–1498.
- Poltavets, V.; Kochetkova, M.; Pitson, S.M.; Samuel, M.S. The Role of the Extracellular Matrix and Its Molecular and Cellular Regulators in Cancer Cell Plasticity. Front. Oncol. 2018, 8, 431.
- Baghban, R.; Roshangar, L.; Jahanban-Esfahlan, R.; Seidi, K.; Ebrahimi-Kalan, A.; Jaymand, M.; Kolahian, S.; Javaheri, T.; Zare, P. Tumor microenvironment complexity and therapeutic implications at a glance. Cell Commun. Signal. 2020, 18, 59.
- Tan, Z.; Xue, H.; Sun, Y.; Zhang, C.; Song, Y.; Qi, Y. The Role of Tumor Inflammatory Microenvironment in Lung Cancer. Front. Pharmacol. 2021, 12, 688625.
- Perillo, B.; Di Donato, M.; Pezone, A.; Di Zazzo, E.; Giovannelli, P.; Galasso, G.; Castoria, G.; Migliaccio, A. ROS in cancer therapy: The bright side of the moon. Exp. Mol. Med. 2020, 52, 192–203.
- Fernandes, C.; Suares, D.; Yergeri, M.C. Tumor Microenvironment Targeted Nanotherapy. Front. Pharmacol. 2018, 9, 1230.
- Altorki, N.K.; Markowitz, G.J.; Gao, D.; Port, J.L.; Saxena, A.; Stiles, B.; McGraw, T.; Mittal, V. The lung microenvironment: An important regulator of tumour growth and metastasis. Nat. Rev. Cancer 2019, 19, 9–31.
- Laflamme, B. Transcriptional cross-talk between tumor and stromal cells. Nat. Genet. 2014, 46, 933.
- Stankovic, B.; Bjørhovde, H.A.K.; Skarshaug, R.; Aamodt, H.; Frafjord, A.; Müller, E.; Hammarström, C.; Beraki, K.; Bækkevold, E.S.; Woldbæk, P.R.; et al. Immune cell composition in human non-small cell lung cancer. Front. Immunol. 2019, 9, 3101.
- Zheng, X.; Hu, Y.; Yao, C. The paradoxical role of tumor-infiltrating immune cells in lung cancer. Intractable Rare Dis. Res. 2017, 6, 234–241.
- Carrasco-Esteban, E.; Domínguez-Rullán, J.A.; Barrionuevo-Castillo, P.; Pelari-Mici, L.; Leaman, O.; Sastre-Gallego, S.; López-Campos, F. Current role of nanoparticles in the treatment of lung cancer. J. Clin. Transl. Res. 2021, 7, 140.
- Mangal, S.; Gao, W.; Li, T.; Zhou, Q. Pulmonary delivery of nanoparticle chemotherapy for the treatment of lung cancers: Challenges and opportunities. Acta Pharmacol. Sin. 2017, 38, 782–797.
- Thakor, A.S.; Gambhir, S.S. Nanooncology: The future of cancer diagnosis and therapy. CA A Cancer J. Clin. 2013, 63, 395–418.
- Gu, F.; Hu, C.; Tai, Z.; Yao, C.; Tian, J.; Zhang, L.; Xia, Q.; Gong, C.; Gao, Y.; Gao, S. Tumour microenvironment-responsive lipoic acid nanoparticles for targeted delivery of docetaxel to lung cancer. Sci. Rep. 2016, 6, 36281.
- Palanikumar, L.; Al-Hosani, S.; Kalmouni, M.; Nguyen, V.P.; Ali, L.; Pasricha, R.; Barrera, F.N.; Magzoub, M. pH-responsive high stability polymeric nanoparticles for targeted delivery of anticancer therapeutics. Commun. Biol. 2020, 3, 1–17.
- Uthaman, S.; Huh, K.M.; Park, I.-K. Tumor microenvironment-responsive nanoparticles for cancer theragnostic applications. Biomater. Res. 2018, 22, 1–11.
- Engelberg, S.; Netzer, E.; Assaraf, Y.G.; Livney, Y.D. Selective eradication of human non-small cell lung cancer cells using aptamer-decorated nanoparticles harboring a cytotoxic drug cargo. Cell Death Dis. 2019, 10, 1–14.
- Babu, A.; Templeton, A.K.; Munshi, A.; Ramesh, R. Nanoparticle-Based Drug Delivery for Therapy of Lung Cancer: Progress and Challenges. J. Nanomater. 2013, 2013, 1–11.
- Siriwon, N.; Bejan, D.; Sherif, P.; Pdx, R.; Wang, R.; Pharmaceuticals, P.; Nelson, M.; Zaidan, H.; Hoang, N.; Bindal, A.; et al. Development of novel immunotherapy based on nanoparticle co-delivering PLK1 and PD-L1 inhibitors for lung cancer treatment Moataz Reda PDX Pharmaceuticals Worapol Ngamcherdtrakul PDX Pharmaceuticals. Preprint 2021.
- Fletcher, J.; Williams, R.T.; Henderson, M.J.; Norris, M.D.; Haber, M. ABC transporters as mediators of drug resistance and contributors to cancer cell biology. Drug Resist. Updat. 2016, 26, 1–9.
- Bar-Zeev, M.; Livney, Y.D.; Assaraf, Y.G. Targeted nanomedicine for cancer therapeutics: Towards precision medicine overcoming drug resistance. Drug Resist. Updat. 2017, 31, 15–30.
- Gillet, J.-P.; Gottesman, M.M. Mechanisms of Multidrug Resistance in Cancer. Methods Mol. Biol. 2010, 596, 47–76.
- Dong, Y.; Feng, S.-S. Poly(d,l-lactide-co-glycolide)/montmorillonite nanoparticles for oral delivery of anticancer drugs. Biomaterials 2005, 26, 6068–6076.
- Peng, S.; Wang, J.; Lu, C.; Xu, Z.; Chai, J.-J.; Ke, Q.; Deng, X.-Z. Emodin enhances cisplatin sensitivity in non-small cell lung cancer through Pgp downregulation. Oncol. Lett. 2021, 21, 1.
- Jang, S.H.; Wientjes, M.G.; Au, J.L. Kinetics of P-glycoprotein-mediated efflux of paclitaxel. J. Pharmacol. Exp. Ther. 2001, 298, 1236–1242.
- Galletti, E.; Magnani, M.; Renzulli, M.L.; Botta, M. Paclitaxel and Docetaxel Resistance: Molecular Mechanisms and Development of New Generation Taxanes. ChemMedChem 2007, 2, 920–942.
- Bergman, A.M.; Pinedo, H.M.; Talianidis, I.; Veerman, G.; Loves, W.J.P.; Van Der Wilt, C.L.; Peters, G.J. Increased sensitivity to gemcitabine of P-glycoprotein and multidrug resistance-associated protein-overexpressing human cancer cell lines. Br. J. Cancer 2003, 88, 1963–1970.
- Berger, W.; Setinek, U.; Hollaus, P.; Zidek, T.; Steiner, E.; Elbling, L.; Cantonati, H.; Attems, J.; Gsur, A.; Micksche, M. Multidrug resistance markers P-glycoprotein, multidrug resistance protein 1, and lung resistance protein in non-small cell lung cancer: Prognostic implications. J. Cancer Res. Clin. Oncol. 2005, 131, 355–363.
- Chen, Z.; Le, H.; Zhang, Y.; Qian, L.; Sekhar, K.R.; Li, W. Lung Resistance Protein and Multidrug Resistance Protein in Non-Small Cell Lung Cancer and Their Clinical Significance. J. Int. Med. Res. 2011, 39, 1693–1700.
- Chen, S.; Bie, M.; Wang, X.; Fan, M.; Chen, B.; Shi, Q.; Jiang, Y. PGRN exacerbates the progression of non-small cell lung cancer via PI3K/AKT/Bcl-2 antiapoptotic signaling. Genes Dis. 2021.
- Ke, W.; Zhao, X.; Lu, Z. Foeniculum vulgare seed extract induces apoptosis in lung cancer cells partly through the down-regulation of Bcl. Biomed. Pharmacother. 2021, 135, 111213.
- Zhang, S.; Liu, N.; Ma, M.; Huang, H.; Handley, M.; Bai, X.; Shan, F. Methionine enkephalin (MENK) suppresses lung cancer by regulating the Bcl-2/Bax/caspase-3 signaling pathway and enhancing natural killer cell-driven tumor immunity. Int. Immunopharmacol. 2021, 98, 107837.
- Tan, X.; Shi, L.; Banerjee, P.; Liu, X.; Guo, H.-F.; Yu, J.; Bota-Rabassedas, N.; Rodriguez, B.L.; Gibbons, D.L.; Russell, W.K.; et al. A protumorigenic secretory pathway activated by p53 deficiency in lung adenocarcinoma. J. Clin. Investig. 2021, 131, 131.
- Zhang, Y.; Han, C.Y.; Duan, F.G.; Fan, X.-X.; Yao, X.-J.; Parks, R.J.; Tang, Y.-J.; Wang, M.-F.; Liu, L.; Tsang, B.K.; et al. p53 sensitizes chemoresistant non-small cell lung cancer via elevation of reactive oxygen species and suppression of EGFR/PI3K/AKT signaling. Cancer Cell Int. 2019, 19, 1–13.
- Gibbons, D.L.; Byers, L.A.; Kurie, J.M. Smoking, p53 Mutation, and Lung Cancer. Mol. Cancer Res. 2014, 12, 3–13.
- Tang, D.; Zhao, X.; Zhang, L.; Wang, Z.; Wang, C. Identification of hub genes to regulate breast cancer metastasis to brain by bioinformatics analyses. J. Cell. Biochem. 2019, 120, 9522–9531.
- Ma, S.; Li, X.; Ran, M.; Ji, M.; Gou, J.; Yin, T.; He, H.; Wang, Y.; Zhang, Y.; Tang, X. Fabricating nanoparticles co-loaded with survivin siRNA and Pt(IV) prodrug for the treatment of platinum-resistant lung cancer. Int. J. Pharm. 2021, 601, 120577.
- Li, B.; Li, Q.; Mo, J.; Dai, H. Drug-Loaded Polymeric Nanoparticles for Cancer Stem Cell Targeting. Front. Pharmacol. 2017, 8, 51.
- Prabavathy, D.; Swarnalatha, Y.; Ramadoss, N. Lung cancer stem cells—Origin, characteristics and therapy. Stem Cell Investig. 2018, 5, 6.
- Maiuthed, A.; Chantarawong, W.; Chanvorachote, P. Lung Cancer Stem Cells and Cancer Stem Cell-targeting Natural Compounds. Anticancer Res. 2018, 38, 3797–3809.
- Masciale, V.; Grisendi, G.; Banchelli, F.; D’Amico, R.; Maiorana, A.; Sighinolfi, P.; Stefani, A.; Morandi, U.; Dominici, M.; Aramini, B. Isolation and Identification of Cancer Stem-Like Cells in Adenocarcinoma and Squamous Cell Carcinoma of the Lung: A Pilot Study. Front. Oncol. 2019, 9, 1394.
- Herreros-Pomares, A.; De-Maya-Girones, J.D.; Calabuig-Fariñas, S.; Lucas, R.; Martínez, A.; Pardo-Sánchez, J.M.; Alonso, S.; Blasco, A.; Guijarro, R.; Martorell, M.; et al. Lung tumorspheres reveal cancer stem cell-like properties and a score with prognostic impact in resected non-small-cell lung cancer. Cell Death Dis. 2019, 10, 1–14.
- Li, Y.; Shi, S.; Ming, Y.; Wang, L.; Li, C.; Luo, M.; Li, Z.; Li, B.; Chen, J. Specific cancer stem cell-therapy by albumin nanoparticles functionalized with CD44-mediated targeting. J. Nanobiotechnol. 2018, 16, 1–15.
- Liu, D.; Hong, Y.; Li, Y.; Hu, C.; Yip, T.-C.; Yu, W.K.; Zhu, Y.; Fong, C.-C.; Wang, W.; Au, S.-K.; et al. Targeted destruction of cancer stem cells using multifunctional magnetic nanoparticles that enable combined hyperthermia and chemotherapy. Theranostics 2020, 10, 1181.
- Di Nicolantonio, F.; Mercer, S.J.; Knight, L.; Gabriel, F.G.; Whitehouse, P.; Sharma, S.; Fernando, A.; Glaysher, S.; Di Palma, S.; Johnson, P.; et al. Cancer cell adaptation to chemotherapy. BMC Cancer 2005, 5, 78.
- Satta, T.; Isobe, K.-I.; Yamauchi, M.; Nakashima, I.; Takagi, H. Expression of MDR1 and glutatione S transferase-π genes and chemosensitivities in human gastrointestinal cancer. Cancer 1992, 69, 941–946.
- Guengerich, F.P. Cytochrome P450 and Chemical Toxicology. Chem. Res. Toxicol. 2008, 21, 70–83.
- Jancova, P.; Anzenbacher, P.; Anzenbacherova, E. Phase II Drug Metabolizing Enzymes. Biomed. Pap. 2010, 154, 103–116.
- Iyanagi, T. Molecular Mechanism of Phase I and Phase II Drug-Metabolizing Enzymes: Implications for Detoxification. Int. Rev. Cytol. 2007, 260, 35–112.
- Patel, M.; Taskar, K.S.; Zamek-Gliszczynski, M.J. Importance of Hepatic Transporters in Clinical Disposition of Drugs and Their Metabolites. J. Clin. Pharmacol. 2016, 56, S23–S39.
- Sau, A.; Tregno, F.P.; Valentino, F.; Federici, G.; Caccuri, A.M. Glutathione transferases and development of new principles to overcome drug resistance. Arch. Biochem. Biophys. 2010, 500, 116–122.
- Tew, K.D.; Gaté, L. Glutathione S-transferases as emerging therapeutic targets. Expert Opin. Ther. Targets 2001, 5, 477–489.
- Chen, C.-S.; Lin, J.T.; Goss, K.A.; He, Y.-A.; Halpert, J.R.; Waxman, D.J. Activation of the Anticancer Prodrugs Cyclophosphamide and Ifosfamide: Identification of Cytochrome P450 2B Enzymes and Site-Specific Mutants with Improved Enzyme Kinetics. Mol. Pharmacol. 2004, 65, 1278–1285.
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death. Cell Death Differ. 2018, 25, 486–541.
- Julien, O.; Wells, J.A. Caspases and their substrates. Cell Death Differ. 2017, 24, 1380–1389.
- Mohammad, R.M.; Muqbil, I.; Lowe, L.; Yedjou, C.; Hsu, H.-Y.; Lin, L.-T.; Siegelin, M.D.; Fimognari, C.; Kumar, N.B.; Dou, Q.P.; et al. Broad targeting of resistance to apoptosis in cancer. Semin. Cancer Biol. 2015, 35, S78–S103.
- Balaji, S.; Terrero, D.; Tiwari, A.K.; Ashby, C.R.; Raman, D. Alternative approaches to overcome chemoresistance to apoptosis in cancer. Adv. Protein Chem. Struct. Biol. 2021, 126, 91–122.
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674.
- Igney, F.H.; Krammer, P.H. Death and anti-death: Tumour resistance to apoptosis. Nat. Cancer 2002, 2, 277–288.
- Czabotar, P.E.; Lee, E.; Thompson, G.V.; Wardak, A.Z.; Fairlie, W.; Colman, P.M. Mutation to Bax beyond the BH3 Domain Disrupts Interactions with Pro-survival Proteins and Promotes Apoptosis. J. Biol. Chem. 2011, 286, 7123–7131.
- Le Gallo, M.; Poissonnier, A.; Blanco, P.; Legembre, P. CD95/Fas, Non-Apoptotic Signaling Pathways, and Kinases. Front. Immunol. 2017, 8, 1216.
- Yuan, X.-J.; Whang, Y. PTEN sensitizes prostate cancer cells to death receptor-mediated and drug-induced apoptosis through a FADD-dependent pathway. Oncogene 2002, 21, 319–327.
- Fulda, S.; Vucic, D. Targeting IAP proteins for therapeutic intervention in cancer. Nat. Rev. Drug Discov. 2012, 11, 109–124.
- Rathore, R.; McCallum, J.E.; Varghese, E.; Florea, A.-M.; Büsselberg, D. Overcoming chemotherapy drug resistance by targeting inhibitors of apoptosis proteins (IAPs). Apoptosis 2017, 22, 898–919.
- Shim, M.K.; Moon, Y.; Yang, S.; Kim, J.; Cho, H.; Lim, S.; Yoon, H.Y.; Seong, J.-K.; Kim, K. Cancer-specific drug-drug nanoparticles of pro-apoptotic and cathepsin B-cleavable peptide-conjugated doxorubicin for drug-resistant cancer therapy. Biomaterials 2020, 261, 120347.
- Byler, S.; Heerboth, S.; Lapinska, K.; Longacre, M.; Snyder, N.; Sarkar, S. Drug resistance in cancer: An overview. Cancers 2014, 6, 1769–1792.
- Lynch, T.J.; Bell, D.W.; Sordella, R.; Gurubhagavatula, S.; Okimoto, R.A.; Brannigan, B.W.; Harris, P.L.; Haserlat, S.M.; Supko, J.G.; Haluska, F.G.; et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N. Engl. J. Med. 2004, 350, 2129–2139.
- Turajlic, S.; Furney, S.; Stamp, G.; Rana, S.; Ricken, G.; Oduko, Y.; Saturno, G.; Springer, C.; Hayes, A.; Gore, M.; et al. Whole-genome sequencing reveals complex mechanisms of intrinsic resistance to BRAF inhibition. Ann. Oncol. 2014, 25, 959–967.
- Dearden, S.; Stevens, J.; Wu, Y.-L.; Blowers, D. Mutation incidence and coincidence in non small-cell lung cancer: Meta-analyses by ethnicity and histology (mutMap). Ann. Oncol. 2013, 24, 2371–2376.
- Kim, S.; Kim, T.M.; Kim, D.-W.; Go, H.; Keam, B.; Lee, S.-H.; Ku, J.-L.; Chung, D.H.; Heo, D.S. Heterogeneity of Genetic Changes Associated with Acquired Crizotinib Resistance in ALK-Rearranged Lung Cancer. J. Thorac. Oncol. 2013, 8, 415–422.
- Mitiushkina, N.V.; Iyevleva, A.G.; Poltoratskiy, A.N.; Ivantsov, A.O.; Togo, A.V.; Polyakov, I.S.; Orlov, S.V.; Matsko, D.E.; Novik, V.I.; Imyanitov, E.N. Detection ofEGFRmutations andEML4-ALKrearrangements in lung adenocarcinomas using archived cytological slides. Cancer Cytopathol. 2013, 121, 370–376.
- Gainor, J.; Dardaei, L.; Yoda, S.; Friboulet, L.; Leischler, I.; Katayama, R.; Dagogo-Jack, I.; Gadgeel, S.; Schultz, K.; Singh, M.; et al. Molecular mechanisms of resistance to first- and second-generation ALK inhibitors in ALK-rearranged lung cancer. Eur. J. Cancer 2016, 69, S138.
- Liu, C.; Shaurova, T.; Shoemaker, S.; Petkovich, M.; Hershberger, P.A.; Wu, Y. Tumor-Targeted Nanoparticles Deliver a Vitamin D-Based Drug Payload for the Treatment of EGFR Tyrosine Kinase Inhibitor-Resistant Lung Cancer. Mol. Pharm. 2018, 15, 3216–3226.
- Ramanathan, S.; Gopinath, S.C.B.; Arshad, M.K.M.; Poopalan, P.; Anbu, P. A DNA based visual and colorimetric aggregation assay for the early growth factor receptor (EGFR) mutation by using unmodified gold nanoparticles. Mikrochim. Acta 2019, 186, 546.
- Helleday, T.; Petermann, E.; Lundin, C.; Hodgson, B.; Sharma, R.A. DNA repair pathways as targets for cancer therapy. Nat. Cancer 2008, 8, 193–204.
- Ulldemolins, A.; Seras-Franzoso, J.; Andrade, F.; Rafael, D.; Abasolo, I.; Gener, P.; Jr, S.S. Perspectives of nano-carrier drug delivery systems to overcome cancer drug resistance in the clinics. Cancer Drug Resist 2021, 4, 44–68.
- Desai, A.; Yan, Y.; Gerson, S.L. Advances in therapeutic targeting of the DNA damage response in cancer. DNA Repair 2018, 66-67, 24–29.
- Rocha, C.R.R.; Silva, M.M.; Quinet, A.; Cabral-Neto, J.B.; Menck, C.F.M. DNA repair pathways and cisplatin resistance: An intimate relationship. Clinics 2018, 73, e478s.
- Yu, W.K.; Wang, Z.; Fong, C.-C.; Liu, D.; Yip, T.-C.; Au, S.-K.; Zhu, G.; Yang, M. Chemoresistant lung cancer stem cells display high DNA repair capability to remove cisplatin-induced DNA damage. Br. J. Pharmacol. 2017, 174, 302–313.
- Olaussen, K.A.; Dunant, A.; Fouret, P.; Brambilla, E.; Andre, F.; Haddad, V.; Taranchon, E.; Filipits, M.; Pirker, R.; Popper, H.H.; et al. DNA Repair by ERCC1 in Non–Small-Cell Lung Cancer and Cisplatin-Based Adjuvant Chemotherapy. N. Engl. J. Med. 2006, 355, 983–991.
- Tell, G.; Damante, G.; Caldwell, D.; Kelley, M.R. The Intracellular Localization of APE1/Ref-1: More than a Passive Phenomenon? Antioxid. Redox Signal. 2005, 7, 367–384.
- Huang, R.; Zhou, P.-K. DNA damage repair: Historical perspectives, mechanistic pathways and clinical translation for targeted cancer therapy. Signal Transduct. Target. Ther. 2021, 6, 254.
- Godin-Heymann, N.; Ulkus, L.; Brannigan, B.W.; McDermott, U.; Lamb, J.; Maheswaran, S.; Settleman, J.; Haber, D.A. The T790M “gatekeeper” mutation in EGFR mediates resistance to low concentrations of an irreversible EGFR inhibitor. Mol. Cancer Ther. 2008, 7, 874–879.
- Sequist, L.V.; Waltman, B.A.; Dias-Santagata, D.; Digumarthy, S.; Turke, A.B.; Fidias, P.; Bergethon, K.; Shaw, A.T.; Gettinger, S.; Cosper, A.K.; et al. Genotypic and Histological Evolution of Lung Cancers Acquiring Resistance to EGFR Inhibitors. Sci. Transl. Med. 2011, 3, 75ra26.
- Engelman, J.A.; Zejnullahu, K.; Mitsudomi, T.; Song, Y.; Hyland, C.; Park, J.O.; Lindeman, N.; Gale, C.-M.; Zhao, X.; Christensen, J.; et al. MET Amplification Leads to Gefitinib Resistance in Lung Cancer by Activating ERBB3 Signaling. Science 2007, 316, 1039–1043.
- Turke, A.B.; Zejnullahu, K.; Wu, Y.-L.; Song, Y.; Dias-Santagata, D.; Lifshits, E.; Toschi, L.; Rogers, A.; Mok, T.; Sequist, L.; et al. Preexistence and Clonal Selection of MET Amplification in EGFR Mutant NSCLC. Cancer Cell 2010, 17, 77–88.
- Sattler, M.; Hasina, R.; Reddy, M.M.; Gangadhar, T.; Salgia, R. The role of the c-Met pathway in lung cancer and the potential for targeted therapy. Ther. Adv. Med Oncol. 2011, 3, 171–184.
- Shi, P.; Oh, Y.-T.; Zhang, G.; Yao, W.; Yue, P.; Li, Y.; Kanteti, R.; Riehm, J.; Salgia, R.; Owonikoko, T.K.; et al. Met gene amplification and protein hyperactivation is a mechanism of resistance to both first and third generation EGFR inhibitors in lung cancer treatment. Cancer Lett. 2016, 380, 494–504.
- Le, X.; Puri, S.; Negrao, M.V.; Nilsson, M.B.; Robichaux, J.; Boyle, T.; Hicks, J.K.; Lovinger, K.L.; Roarty, E.; Rinsurongkawong, W.; et al. Landscape of EGFR-Dependent and -Independent Resistance Mechanisms to Osimertinib and Continuation Therapy Beyond Progression in EGFR-Mutant NSCLC. Clin. Cancer Res. 2018, 24, 6195–6203.
- Yabuki, N.; Sakata, K.; Yamasaki, T.; Terashima, H.; Mio, T.; Miyazaki, Y.; Fujii, T.; Kitada, K. Gene amplification and expression in lung cancer cells with acquired paclitaxel resistance. Cancer Genet. Cytogenet. 2007, 173, 1–9.
- Chen, C.-J.; Chin, J.E.; Ueda, K.; Clark, D.P.; Pastan, I.; Gottesman, M.M.; Roninson, I.B. Internal duplication and homology with bacterial transport proteins in the mdr1 (P-glycoprotein) gene from multidrug-resistant human cells. Cell 1986, 47, 381–389.
- Gottesman, M.M.; Fojo, T.; Bates, S.E. Multidrug resistance in cancer: Role of ATP–dependent transporters. Nat. Cancer 2002, 2, 48–58.
- Saad, M.; Garbuzenko, O.B.; Minko, T. Co-delivery of siRNA and an anticancer drug for treatment of multidrug-resistant cancer. Nanomedicine 2008, 3, 761–776.
- Zhu, B.; Reinberg, D. Epigenetic inheritance: Uncontested? Cell Res. 2011, 21, 435–441.
- Roberti, A.; Valdes, A.F.; Torrecillas, R.; Fraga, M.F.; Fernandez, A.F. Epigenetics in cancer therapy and nanomedicine. Clin. Epigenet. 2019, 11, 81.
- Cheng, Y.; He, C.; Wang, M.; Ma, X.; Mo, F.; Yang, S.; Han, J.; Wei, X. Targeting epigenetic regulators for cancer therapy: Mechanisms and advances in clinical trials. Signal Transduct. Target. Ther. 2019, 4, 62.
- Wilting, R.H.; Dannenberg, J.-H. Epigenetic mechanisms in tumorigenesis, tumor cell heterogeneity and drug resistance. Drug Resist. Updat. 2012, 15, 21–38.
- Glozak, M.; Seto, E. Histone deacetylases and cancer. Oncogene 2007, 26, 5420–5432.
- Mamdani, H.; Jalal, S.I. Histone Deacetylase Inhibition in Non-small Cell Lung Cancer: Hype or Hope? Front. Cell Dev. Biol. 2020, 8, 582370.
- Wang, L.; Li, H.; Ren, Y.; Zou, S.; Fang, W.; Jiang, X.; Jia, L.; Li, M.; Liu, X.; Yuan, X.; et al. Targeting HDAC with a novel inhibitor effectively reverses paclitaxel resistance in non-small cell lung cancer via multiple mechanisms. Cell Death Dis. 2016, 7, e2063.
- Sharma, S.V.; Lee, D.Y.; Li, B.; Quinlan, M.P.; Takahashi, F.; Maheswaran, S.; McDermott, U.; Azizian, N.; Zou, L.; Fischbach, M.A.; et al. A Chromatin-Mediated Reversible Drug-Tolerant State in Cancer Cell Subpopulations. Cell 2010, 141, 69–80.
- Wang, E.C.; Min, Y.; Palm, R.C.; Fiordalisi, J.J.; Wagner, K.T.; Hyder, N.; Cox, A.D.; Caster, J.; Tian, X.; Wang, A.Z. Nanoparticle formulations of histone deacetylase inhibitors for effective chemoradiotherapy in solid tumors. Biomaterials 2015, 51, 208–215.
- Tran, T.H.; Ramasamy, T.; Truong, D.H.; Shin, B.S.; Choi, H.-G.; Yong, C.S.; Kim, J.O. Development of Vorinostat-Loaded Solid Lipid Nanoparticles to Enhance Pharmacokinetics and Efficacy against Multidrug-Resistant Cancer Cells. Pharm. Res. 2014, 31, 1978–1988.
- Green, M.R.; Manikhas, G.M.; Orlov, S.; Afanasyev, B.; Makhson, A.M.; Bhar, P.; Hawkins, M.J. Abraxane®, a novel Cremophor®-free, albumin-bound particle form of paclitaxel for the treatment of advanced non-small-cell lung cancer. Ann. Oncol. 2006, 17, 1263–1268.