Human pluripotent stem cells (hPSCs) are receiving much attention in the field of tissue regeneration. They can be differentiated into any cell type in the human body and subsequently developed into heterogeneous tissues or organs in vitro for implantation. Conventionally, hPSCs are harvested from surplus embryos that were generated after in vitro fertilization, known as embryonic stem cells (ESCs). This, however, comes with inherent ethical and practical issues which limit their use in regenerative medicine.
The keystone discovery of induced pluripotent stem cell (iPSC) technology provides an alternative and endless source, circumventing the unfavorable issues with embryonic stem cells, and yielding fundamental advantages.
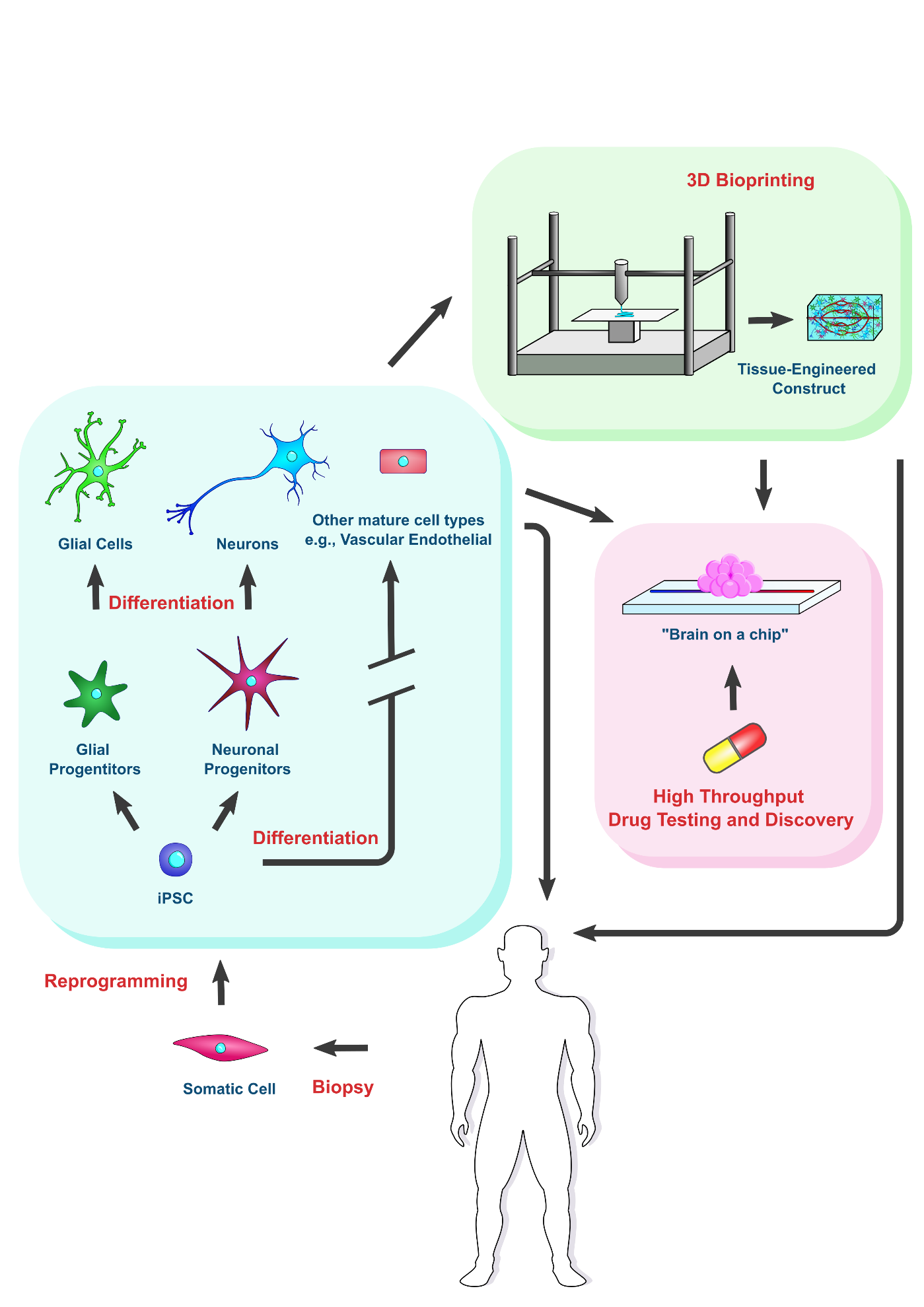
Pararkinson's and Alzheimer's disease are types of neurodegenrative conditions resulting in which the build up of harmful protein aggregates leads to neural cell death. IPSC technology offers promise for in vitro modeling pathophysiology and the treatment of these diseases. Additionally, the rapidly evolving field of 3D bioprinting used in conjunction iPSC-based tissue engineering offers new prospects for both disease modeling and treatment.
- induced pluripotent stem cells
- Alzheimer’s disease
- Parkinson’s disease
- neural cells
- neural organoids
- 3D bioprinting
1. Prospect of Human Pluripotent Stem Cells
2. iPSCs for Regenerative Cell Therapies Applied to Alzheimer’s and Parkinson’s Treatment
| Source of iPSCs |
Neurodegenerative Disease Treated |
Model | Type of Cells | Number | Route of Delivery |
Outcome | Reference |
|---|---|---|---|---|---|---|---|
| Autologous, mouse skin fibroblasts | AD | in vivo: 5XFAD mice | iPSCs | 100,000 | Injection into subiculum |
Decrease in Aβ plaque deposition and beta/gamma-secretase activity |
[29] |
| Autologous, skin fibroblasts | PD | in vivo: Parkinsonian cynomolgus monkeys | Dopaminergic neurons | 10–40 million | Injection into four sites of post- commissural putamen |
Improvements in motor function and reinnervation by implanted neurons | [31] |
| Autologous and allogeneic, skin fibroblasts | PD | in vivo: Parkinsonian rhesus monkeys | Dopaminergic neurons | 5.5–22 million | Injection into basal ganglia |
Improvements in motor function consistent with reinnervation by implanted neurons seen in autologous transplant group |
[32] |
| Human dermal fibroblast lines | PD | in vivo: immunodeficient 6-OHDA Parkinsonian mice | Dopaminergic neuron progenitors | 100,000–300,000 | Injection into striatum |
Recovery of rotation behavior, improvements on corridor, cylinder, stepping tests |
[33] |
3. Using iPSC-Derived Organoids to Model Pathophysiology of Neurodegenerative Diseases
4. Utility of Bioprinter for Making Tissue Constructs
A tissue-engineered construct can be implanted to replace damaged tissue or used to
accurately model the disease environment, lending itself to drug discovery and testing [61][46].To engineer a tissue construct, three components are typically required: cells, dissolved signaling molecules, and a scaffold [62][47]. Patient-specific iPSCs are ideal candidates for building a tissue construct for pathological modelling or implantation. Dissolved signaling molecules around the cells are crucial for guiding cell behavior, and therefore the response of implanted cells to dissolved factors in situ should be carefully considered. An implanted construct could be supplemented with additional signaling molecules to guide cell fate in vivo. Cells also respond to physical and mechanical cues in their surroundings, naturally conferred by the ECM in vivo.
References
- Mehat, M.S.; Sundaram, V.; Ripamonti, C.; Robson, A.G.; Smith, A.J.; Borooah, S.; Robinson, M.; Rosenthal, A.N.; Innes, W.; Weleber, R.G.; et al. Transplantation of Human Embryonic Stem Cell-Derived Retinal Pigment Epithelial Cells in Macular Degeneration. Ophthalmology 2018, 125, 1765–1775.
- Schwartz, S.D.; Hubschman, J.-P.; Heilwell, G.; Franco-Cardenas, V.; Pan, C.; Ostrick, R.M.; Mickunas, E.; Gay, R.; Klimanskaya, I.; Lanza, R. Embryonic stem cell trials for macular degeneration: A preliminary report. Lancet 2012, 379, 713–720.
- Schwartz, S.D.; Regillo, C.D.; Lam, B.L.; Eliott, D.; Rosenfeld, P.J.; Gregori, N.Z.; Hubschman, J.-P.; Davis, J.L.; Heilwell, G.; Spirn, M.; et al. Human embryonic stem cell-derived retinal pigment epithelium in patients with age-related macular degeneration and Stargardt’s macular dystrophy: Follow-up of two open-label phase 1/2 studies. Lancet 2015, 385, 509–516.
- Da Cruz, L.; Fynes, K.; Georgiadis, O.; Kerby, J.; Luo, Y.H.; Ahmado, A.; Vernon, A.; Daniels, J.T.; Nommiste, B.; Hasan, S.M.; et al. Phase 1 clinical study of an embryonic stem cell–derived retinal pigment epithelium patch in age-related macular degeneration. Nat. Biotechnol. 2018, 36, 328–337.
- Menasché, P.; Vanneaux, V.; Hagège, A.; Bel, A.; Cholley, B.; Parouchev, A.; Cacciapuoti, I.; Al-Daccak, R.; Benhamouda, N.; Blons, H.; et al. Transplantation of Human Embryonic Stem Cell–Derived Cardiovascular Progenitors for Severe Ischemic Left Ventricular Dysfunction. J. Am. Coll. Cardiol. 2018, 71, 429–438.
- Wu, J.; Zhou, X.; Tan, Y.; Wang, L.; Li, T.; Li, Z.; Gao, T.; Fan, J.; Guo, B.; Li, W.; et al. Phase 1 trial for treatment of COVID-19 patients with pulmonary fibrosis using hESC-IMRCs. Cell Prolif. 2020, 53, e12944.
- Freed, C.R.; Greene, P.E.; Breeze, R.E.; Tsai, W.-Y.; DuMouchel, W.; Kao, R.; Dillon, S.; Winfield, H.; Culver, S.; Trojanowski, J.Q.; et al. Transplantation of Embryonic Dopamine Neurons for Severe Parkinson’s Disease. N. Engl. J. Med. 2001, 344, 710–719.
- Kim, H.J.; Cho, K.R.; Jang, H.; Lee, N.K.; Jung, Y.H.; Kim, J.P.; Lee, J.I.; Chang, J.W.; Park, S.; Kim, S.T.; et al. Intracerebroventricular injection of human umbilical cord blood mesenchymal stem cells in patients with Alzheimer’s disease dementia: A phase I clinical trial. Alzheimers Res. Ther. 2021, 13, 154.
- Yu, J.; Thomson, J.A. Pluripotent stem cell lines. Genes Dev. 2008, 22, 1987–1997.
- Kelly, J. Practical and Ethical Issues Limiting the Clinical Use of Human Embryonic Stem Cells. Arch. Stem. Cell Res. 2017, 4, 1018.
- Romito, A.; Cobellis, G. Pluripotent Stem Cells: Current Understanding and Future Directions. Stem Cells Int. 2016, 2016, 9451492.
- Semechkin, R.; Isaev, D.; Abramihina, T.; Turovets, N.; West, R.A.; Zogovic-Kapsalis, T.; Semechkin, A. 100. Human Neural Stem Cells of Parthenogenetic Origin. Mol. Ther. 2011, 19 (Suppl. 1), S40.
- Daughtry, B.; Mitalipov, S. Concise Review: Parthenote Stem Cells for Regenerative Medicine: Genetic, Epigenetic, and Developmental Features. Stem Cells Transl. Med. 2014, 3, 290–298.
- Wolf, D.P.; Morey, R.; Kang, E.; Ma, H.; Hayama, T.; Laurent, L.C.; Mitalipov, S. Concise Review: Embryonic Stem Cells Derived by Somatic Cell Nuclear Transfer: A Horse in the Race? Stem Cells 2016, 35, 26–34.
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676.
- Choi, J.; Lee, S.; Mallard, W.; Clement, K.; Tagliazucchi, G.M.; Lim, H.; Choi, I.Y.; Ferrari, F.; Tsankov, A.M.; Pop, R.; et al. A comparison of genetically matched cell lines reveals the equivalence of human iPSCs and ESCs. Nat. Biotechnol. 2015, 33, 1173–1181.
- Puri, M.C.; Nagy, A. Concise Review: Embryonic Stem Cells Versus Induced Pluripotent Stem Cells: The Game Is On. Stem Cells 2011, 30, 10–14.
- Bi, H.; Karanth, S.S.; Ye, K.; Stein, R.; Jin, S. Decellularized Tissue Matrix Enhances Self-Assembly of Islet Organoids from Pluripotent Stem Cell Differentiation. ACS Biomater. Sci. Eng. 2020, 6, 4155–4165.
- Bi, H.; Ye, K.; Jin, S. Proteomic analysis of decellularized pancreatic matrix identifies collagen V as a critical regulator for islet organogenesis from human pluripotent stem cells. Biomaterials 2020, 233, 119673.
- Karanth, S.S.; Sun, S.; Bi, H.; Ye, K.; Jin, S. Angiopoietins stimulate pancreatic islet development from stem cells. Sci. Rep. 2021, 11, 13558.
- Marei, H.E.; Althani, A.; Lashen, S.; Cenciarelli, C.; Hasan, A. Genetically unmatched human iPSC and ESC exhibit equivalent gene expression and neuronal differentiation potential. Sci. Rep. 2017, 7, 17504.
- Goto, K.; Imamura, K.; Komatsu, K.; Mitani, K.; Aiba, K.; Nakatsuji, N.; Inoue, M.; Kawata, A.; Yamashita, H.; Takahashi, R.; et al. Simple Derivation of Spinal Motor Neurons from ESCs/iPSCs Using Sendai Virus Vectors. Mol. Ther. —Methods Clin. Dev. 2017, 4, 115–125.
- Wen, Y.; Jin, S. Production of neural stem cells from human pluripotent stem cells. J. Biotechnol. 2014, 188, 122–129.
- Zhang, F.; Qiu, H.; Dong, X.; Wang, C.; Na, J.; Zhou, J.; Wang, C. Transferrin improved the generation of cardiomyocyte from human pluripotent stem cells for myocardial infarction repair. J. Mol. Histol. 2020, 52, 87–99.
- Kumar, S.V.; Er, P.X.; Lawlor, K.; Motazedian, A.; Scurr, M.; Ghobrial, I.; Combes, A.N.; Zappia, L.; Oshlack, A.; Stanley, E.G.; et al. Kidney micro-organoids in suspension culture as a scalable source of human pluripotent stem cell-derived kidney cells. Development 2019, 146, dev172361.
- Hu, B.-Y.; Weick, J.P.; Yu, J.; Ma, L.-X.; Zhang, X.-Q.; Thomson, J.A.; Zhang, S.-C. Neural differentiation of human induced pluripotent stem cells follows developmental principles but with variable potency. Proc. Natl. Acad. Sci. USA 2010, 107, 4335–4340.
- Burke, E.E.; Chenoweth, J.G.; Shin, J.H.; Collado-Torres, L.; Kim, S.-K.; Micali, N.; Wang, Y.; Colantuoni, C.; Straub, R.E.; Hoeppner, D.J.; et al. Dissecting transcriptomic signatures of neuronal differentiation and maturation using iPSCs. Nat. Commun. 2020, 11, 462.
- Halevy, T.; Urbach, A. Comparing ESC and iPSC-Based Models for Human Genetic Disorders. J. Clin. Med. 2014, 3, 1146–1162.
- Cha, M.-Y.; Kwon, Y.-W.; Ahn, H.-S.; Jeong, H.; Lee, Y.Y.; Moon, M.; Baik, S.H.; Kim, D.K.; Song, H.; Yi, E.C.; et al. Protein-Induced Pluripotent Stem Cells Ameliorate Cognitive Dysfunction and Reduce Aβ Deposition in a Mouse Model of Alzheimer’s Disease. STEM CELLS Transl. Med. 2017, 6, 293–305.
- Cho, H.-J.; Lee, C.-S.; Kwon, Y.-W.; Paek, J.S.; Lee, S.-H.; Hur, J.; Lee, E.J.; Roh, T.-Y.; Chu, I.-S.; Leem, S.-H.; et al. Induction of pluripotent stem cells from adult somatic cells by protein-based reprogramming without genetic manipulation. Blood 2010, 116, 386–395.
- Hallett, P.; Deleidi, M.; Astradsson, A.; Smith, G.; Cooper, O.; Osborn, T.M.; Sundberg, M.; Moore, M.A.; Perez-Torres, E.; Brownell, A.-L.; et al. Successful Function of Autologous iPSC-Derived Dopamine Neurons following Transplantation in a Non-Human Primate Model of Parkinson’s Disease. Cell Stem Cell 2015, 16, 269–274.
- Tao, Y.; Vermilyea, S.C.; Zammit, M.; Lu, J.; Olsen, M.; Metzger, J.M.; Yao, L.; Chen, Y.; Phillips, S.; Holden, J.E.; et al. Autologous transplant therapy alleviates motor and depressive behaviors in parkinsonian monkeys. Nat. Med. 2021, 27, 632–639.
- Song, B.; Cha, Y.; Ko, S.; Jeon, J.; Lee, N.; Seo, H.; Park, K.-J.; Lee, I.-H.; Lopes, C.; Feitosa, M.; et al. Human autologous iPSC–derived dopaminergic progenitors restore motor function in Parkinson’s disease models. J. Clin. Investig. 2019, 130, 904–920.
- Schweitzer, J.S.; Song, B.; Herrington, T.M.; Park, T.-Y.; Lee, N.; Ko, S.; Jeon, J.; Cha, Y.; Kim, K.; Li, Q.; et al. Personalized iPSC-Derived Dopamine Progenitor Cells for Parkinson’s Disease. N. Engl. J. Med. 2020, 382, 1926–1932.
- Ford, E.; Pearlman, J.; Ruan, T.; Manion, J.; Waller, M.; Neely, G.G.; Caron, L. Human Pluripotent Stem Cells-Based Therapies for Neurodegenerative Diseases: Current Status and Challenges. Cells 2020, 9, 2517.
- Li, L.; Lin, H.; Hua, P.; Yan, L.; Dong, H.; Li, T.; Liu, W. Polymorphism of the Dopa-Decarboxylase Gene Modifies the Motor Response to Levodopa in Chinese Patients with Parkinson’s Disease. Front. Neurol. 2020, 11, 520934.
- Drozdzik, M.; Bialecka, M.; Kurzawski, M. Pharmacogenetics of Parkinson’s disease—through mechanisms of drug actions. Curr. Genom. 2013, 14, 568–577.
- De Beaumont, L.; Pelleieux, S.; Lamarre-Théroux, L.; Dea, D.; Poirier, J. Butyrylcholinesterase K and Apolipoprotein E-ε4 Reduce the Age of Onset of Alzheimer’s Disease, Accelerate Cognitive Decline, and Modulate Donepezil Response in Mild Cognitively Impaired Subjects. J. Alzheimers Dis. 2016, 54, 913–922.
- Potjewyd, G.; Moxon, S.; Wang, T.; Domingos, M.; Hooper, N.M. Tissue Engineering 3D Neurovascular Units: A Biomaterials and Bioprinting Perspective. Trends Biotechnol. 2018, 36, 457–472.
- Jagadeesan, S.; Workman, M.J.; Herland, A.; Svendsen, C.N.; Vatine, G.D. Generation of a Human iPSC-Based Blood-Brain Barrier Chip. J. Vis. Exp. 2020, 157, e60925.
- Oliveira, L.M.A.; Lockhart, L.J.F.; Botelho, M.G.; Lin, K.-H.; Wales, P.; Koch, J.C.; Gerhardt, E.; Taschenberger, H.; Outeiro, T.F.; Lingor, P.; et al. Elevated α-synuclein caused by SNCA gene triplication impairs neuronal differentiation and maturation in Parkinson’s patient-derived induced pluripotent stem cells. Cell Death Dis. 2015, 6, e1994.
- Mohamed, N.-V.; Sirois, J.; Ramamurthy, J.; Mathur, M.; Lépine, P.; Deneault, E.; Maussion, G.; Nicouleau, M.; Chen, C.X.-Q.; Abdian, N.; et al. Midbrain organoids with an SNCA gene triplication model key features of synucleinopathy. Brain Commun. 2021, 3, fcab223.
- Raja, W.K.; Mungenast, A.E.; Lin, Y.-T.; Ko, T.; Abdurrob, F.; Seo, J.; Tsai, L.-H. Self-Organizing 3D Human Neural Tissue Derived from Induced Pluripotent Stem Cells Recapitulate Alzheimer’s Disease Phenotypes. PLoS ONE 2016, 11, e0161969.
- Mohamed, N.-V.; Lépine, P.; Lacalle-Aurioles, M.; Sirois, J.; Mathur, M.; Reintsch, W.; Beitel, L.K.; Fon, E.A.; Durcan, T.M. Microfabricated disk technology: Rapid scale up in midbrain organoid generation. Methods 2021.
- Pham, M.T.; Pollock, K.M.; Rose, M.D.; Cary, W.A.; Stewart, H.R.; Zhou, P.; Nolta, J.; Waldau, B. Generation of human vascularized brain organoids. Neuroreport 2018, 29, 588–593.
- Warren, D.; Tomaskovic-Crook, E.; Wallace, G.G.; Crook, J.M. Engineering in vitro human neural tissue analogs by 3D bioprinting and electrostimulation. APL Bioeng. 2021, 5, 020901.
- Romanazzo, S.; Nemec, S.; Roohani, I. iPSC Bioprinting: Where are We at? Materials 2019, 12, 2453.