Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Sara De Salvo and Version 2 by Conner Chen.
Approximately 40% of the skeletal and musculoskeletal proliferative lesions do not determine metastasis and are related to a good survival rate. These groups of proliferative lesions are generally defined as benign bone tumors (BBTs).
- benign bone tumors
- nonmalignant bone tumors
- proliferative bone lesions
1. Introduction
Approximately 40% of the skeletal and musculoskeletal proliferative lesions do not determine metastasis and are related to a good survival rate [1]. These groups of proliferative lesions are generally defined as benign bone tumors (BBTs). However, they can occur in any part of the skeleton and can still be dangerous as they may grow and compress healthy tissues.
This definition includes a wide variety of diseases, which vary in terms of incidence, clinical presentation, and possess a diverse array of therapeutic options. Many classifications have been proposed. The last WHO classification considers the biological behavior of the lesions, dividing benign bone tumors into “intermediate (locally aggressive)” and “intermediate (very rarely metastatic)”. Other classifications are based on the type of matrix production or otherwise histological, clinical, and radiological criteria.
The most frequent BBTs are osteochondroma, osteoma, osteoid osteoma, osteoblastoma, giant cell tumor, aneurysmal bone cyst, fibrous dysplasia, and enchondroma. These tumors are further divided into categories based on their cell type: bone-forming, cartilage-forming, as well as connective tissue and vascular [2][3]. The incidence of BBTs is hard to calculate since they are often asymptomatic and difficult to detect [2]. Some forms of BBTs are sporadic, and due to their low incidence, they will not be discussed.
2. Osteoid Osteoma
2.1. Introduction
A bone-forming tumor is a benign osteoblastic tumor, accounting for 12% of benign tumors of the bone [4]. It can develop at any age, even before 3 years old [5][6], but its typical presentation is in the second decade, more frequently in boys (two to four times more than girls) [7]. Usually solitary, there have been reports about rare metachronous presentations [8]. It is composed of a nidus surrounded by sclerotic tissue whose dimensions are generally less than 1.5 cm in diameter. If the lesion has a longer diameter, it can be considered an osteoblastoma, similar in genetics and morphology [9]. The debate between the differentiation of these two separate entities is still going on in the scientific community.
2.2. Sites of Development
Bone-forming tumors mainly affect lower limb long bones, but a frequent site of presentation is the spine, especially the lumbar district [10].
2.3. Micro-Macroscopic Features
The histopathological exam shows a central nidus composed of tiny osteoid islands lined by osteoblasts. Pain is attributed to unmyelinated nerve fibers present within the nidus [11]. The area peripheral to the nidus appears clearer because of osteoclastic resorption, and dense sclerotic tissue surrounds the nidus. According to some studies, the nidus produces prostaglandins, and it may also produce osteocalcin [12][13]. Its genetic features are similar to those of osteoblastoma; in fact, osteoid osteoma expresses Runx2 and Osterix, transcription factors involved in osteoblastic differentiation [14].
2.4. Clinical Features
Bone-forming tumors present with increasing pain that may worsen at night. One of their main characteristics is that this pain is resolved with NSAIDs (prostaglandin inhibitors) in less than half an hour: if this does not occur, it is very likely not to be an osteoid osteoma and other options have to be investigated [15]. Moreover, bone-forming tumors can present with swelling, erythema, tenderness, and muscular atrophy [16]. The clinical symptomatology may differ based on the presentation site. In patients affected by a spine osteoid osteoma, this can result in scoliotic posture; therefore, differential diagnosis is fundamental [17]. This lesion may affect open physis, causing lengthening or angular deformity of the affected bone. Furthermore, if it affects the hip joint, causing hip impingement; therefore, an accurate evaluation of the femoroacetabular impingement becomes mandatory. When developing intra-articular or juxta-articular, the lesion can cause synovitis [18].
2.5. Imaging
In X-ray examination, a bone-forming tumor appears as a nidus surrounded by sclerotic tissue. It may be hard to identify when it is intra-articular, therefore requiring further look examination. The second-level exam is a CT scan, which displays a small, defined nidus surrounded by a sclerotic reaction which may contain calcifications. Bone scintigraphy usually shows arterial phase uptake in the nidus due to its high vascular concentration and lower concentration in the reactive surrounding bone. There is a typical sign called the double-density sign which is diagnostic. It is fundamental to pay attention to growth plates since they can obscure the signal if the osteoid osteoma is in their proximity; therefore, it can be helpful to evaluate the contralateral side. Other imaging techniques may help the diagnostic workup, but those must be considered case by case [19][20][21] (Figure 1).
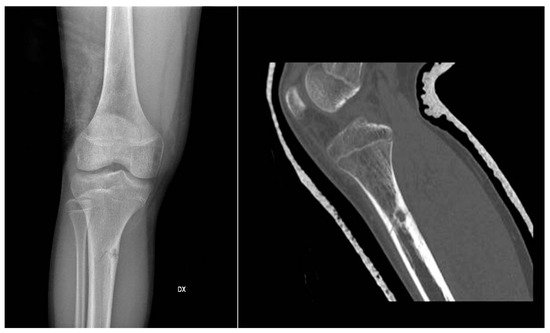
Figure 1.
X-rays and CT scan of a tibial fracture that occurred in the development site of an Osteoid osteoma.
2.6. Differential Diagnosis
For differential diagnosis, it is essential to exclude osteoblastoma that presents with no pain at all or dull pain, responding less to NSAIDs, and it is usually more extensive than the osteoid osteoma and with a more complex morphology. Osteoid osteoma presentation can resemble that of subacute or chronic osteomyelitis, but those typically extend from growth plates.
2.7. Treatment and Prognosis
The natural history of the tumor is of spontaneous resolution within 6 to 15 years, and, if not symptomatic, watchful waiting is the suggested approach. On the other hand, NSAIDs are the drug of choice if symptoms are present. Studies have reported that treatment with NSAIDs or aspirin could reduce the time of healing by 2 or 3 years. In case of symptomatic lesions, such as those causing scoliosis, length discrepancy, and other morpho-functional disorders, those can be approached with surgical resection, radiofrequency, cryotherapy, and MRI-guided high-intensity focused ultrasound. It is not necessary to excise the sclerotic margins; the focus of the resection must be the removal of the nidus. Recurrences can happen if, after resection, the nidus is not entirely excised [15][22][23][24].
3. Osteoblastoma
3.1. Introduction
Accounting for approximately 14% of benign bone tumors, osteoblastoma is a bone-forming neoplasm that affects primarily young adults (mean age 20 years), predominantly males (two to three times more than females). The tumor is more aggressive as the age of presentation is [25]. Like osteoid osteomas, they are composed of a central nidus surrounded by sclerotic tissue. Although the nidus is more vascularized and the lesions less organized, it does not produce prostaglandins, and it carries a lower amount of nerve fibers. The average size is around 3 cm, but it can grow to 15 cm of diameter [26].
3.2. Sites of Development
Osteoblastoma most frequently arises from the posterior elements of the spine and the sacrum. Long tubular bones of the lower limb are another frequent site of presentation with the mandible, and in this last location, it is referred to as cementoblastoma [7][27].
3.3. Micro-Macroscopic Features
An osteoblastoma is a rich vascularized lesion in red/brown color. It is usually sharply distinguished from the bone marrow by a border, and the margins on the cortical surface push against it. Usually, they are separated from other bone structures by a fibrous tissue layer. The borders permeate more as the tumor becomes more aggressive. An essential feature of this lesion and OO is the formation of immature and osteoid bone trabeculae [28]. It is possible to differentiate osteoblastoma from osteosarcoma immunohistochemically because the former presents with a beta-catenin nuclear staining, while the latter presents with a cytoplasmic or membranous one [28]. As mentioned for osteoid osteoma, osteoblastoma is characterized by genetic mutations of FOS and FOSB [29].
3.4. Clinical Features
As with most benign tumors, osteoblastoma tends to grow slowly and be asymptomatic for most patients: this clinical feature may help the differential diagnosis with osteoid osteoma. When present, symptoms may include dull, localized pain, with soft tissue swelling. As in other cases, symptoms may vary based on the presentation site. It can cause nerve root compression, postural scoliosis, and muscle weakness when affecting the spine. Patients with lower extremity affection may present with limp [30][31]. Toxic osteoblastoma is a rare variant of this neoplasm with systemic symptoms, such as fever, anorexia, and diffuse periostitis [32].
3.5. Imaging
The evaluation algorithm of osteoblastoma includes plain radiographs, CT scans, and MRI. Its appearance in plain radiographs can have different patterns. It can be similar to osteoid osteoma but greater (>2 cm) and surrounded by less reactive sclerosis. Benign periosteal reaction is a standard feature. Lesions developing in the axial skeleton can mimic ABCs. Furthermore, mainly aggressive osteoblastomas can resemble malignant lesions when more extensive in size (>4 cm), causing disruption and cortical thinning.
3.6. Differential Diagnosis
The differential diagnosis includes many pathologies, such as osteoid osteoma, osteomyelitis, stress fracture, osteosarcoma, giant cell tumor of bone, and aneurysmal bone cyst. To correctly identify the disease, advanced radiographic imaging might be necessary [33][34].
3.7. Treatment and Prognosis
Differently from osteoid osteoma, osteoblastoma may have an aggressive development: for this reason, it is preferably treated surgically. The procedure of choice varies based on the aggressiveness grade of the lesion. The most frequent options are curettage and resection en bloc, depending on the tumor’s aggressiveness. Intralesional curettage has a higher rate of recurrences, but it is less invasive, so it is used if the suspicion of malignancy or evolution toward it is low. On the other hand, en bloc resection leads to a lower rate of recurrences than other techniques; therefore, it is used in more aggressive lesions. Other therapeutic tools, such as radiotherapy or chemotherapy, have not proven effective for this tumor. It is fundamental to continue the patient follow-up after surgery with imaging exams, to exclude eventual recurrences. If untreated and enlarging, an osteoblastoma can damage the adjacent bone or nervous structures [35][36][37].
4. Giant Cell Tumor
4.1. Introduction
Giant cell tumors represent 15–20% of benign bone neoplasms in the United States, having even higher incidence rates in other countries, such as China [38]. Unlike other benign tumors, they can result metastases (pulmonary in most cases) and are locally aggressive, causing osteolysis. It affects primarily young adults around the third decade of life [39][40].
4.2. Sites of Development
Giant cell tumors frequently develop from the meta-epiphyses of the long bones, especially all four bones that make up the knee joint, particularly the proximal tibia and distal femur [41][42].
4.3. Micro-Macroscopic Features
In the histopathological exam, GCTB is friable and of red/brown color. The giant cells are multinucleated and immersed in a stromal background of monocytes and spindle cells. The pattern can be variable, including hemosiderin, necrosis, and hemorrhage. The neoplastic cells are the mononuclear spindle cells. [43] Studies on its genetics have shown that 20% of GCTB have p53 overexpressed, and those are associated with neoplasm recurrence and metastases [44]. Other findings have been increased telomerase activity and their shortening prevention [45]. A study found that 54% of GCTB had a 20q11 amplification [46]. RANKL overexpression is the molecular feature most represented in GCTB, particularly in osteoblast-like mononuclear stromal cells, recruiting osteoclast cells. The latter would then cause osteolysis. Histone modification, found in over 90% of these tumors, may be one of the starting points of neoplastic formation [47][48].
4.4. Clinical Features
Patients typically present with pain. At physical examination, it can be possible to find tenderness to palpation, swelling, and, in case of joint proximity, joint effusion. Antalgic gait may also be a frequent finding. Pathologic fractures can be an indirect sign of the presence of the lesion, often involving the joints [49].
4.5. Imaging
The imaging workup comprehends plain X-rays, CT scan, and MRI exams. Simple X-rays can show various patterns, typically a radiolucent meta-epiphyseal lesion. One of the most used classifications was created by Campanacci in 1987 (Table 1).
Table 1.
Campanacci Grading.
| Grade | Radiological Finding |
|---|---|
| I | Lesions confined in the bone cortex, which is intact (cystic lesions) |
| II | Lesions confined in the bone cortex, which appears thinned but not perforated |
| III | Lesions extended over the bone, that invade the cortex and expand to the soft tissues |
This classification was made to influence the extent of surgery. Even if it simplifies the diagnostic workup, other histopathological and genetic factors must be evaluated to clarify the tumor’s aggressiveness. CT can be used to define the lesion and its borders further. MRI findings are often nonspecific, with the T1-weighted sequence showing a decrease in signal intensity and increase in the T2-weighted signal: these sequences can also spot hemosiderin deposition, a frequent finding in GCTB, indicated by low signal intensity. Another nonspecific but frequent feature of GCTB in scintigraphy is the “donut sign” due to the intense uptake of radionuclide in the periphery and the necrosis in the center of the lesion. Chest CT imaging is suggested in recurrent GCTB to spot eventual pulmonary metastases [50][51].
4.6. Differential Diagnosis
Differential diagnosis includes benign and malignant tumors rich in giant cells and osteoclasts, such as brown tumors associated with hyperparathyroidism and metastases of other body tumors [52].
4.7. Treatment
The gold standard for GCTB is surgery. A frequent approach consists of intralesional curettage and cavity filling with PMMA (polymethylmethacrylate). Extensive bone resection is typically performed if the tumor is located in expendable bones such as fibula or distal ulna. In the case of extraosseous extension and metastases, different approaches, such as denosumab, radiotherapy, bisphosphonates, and chemotherapy, can be evaluated. There is no consensus about the follow-up, but these patients must be periodically checked on with imaging exams [53][54][55][56]. Metastases affect 2–3% of GCTB and develop almost exclusively from the lungs (especially if the primary tumor arises from the spine). These metastases though, maintain the histological and morphological pattern of the tumor of origin, and therefore are often referred to as “benign” pulmonary implants. These are most frequently found in recurrent lesions [57][58].
References
- Harrleson, J. Bone Tumors: General Aspects and Data on 6221 Cases. Ann. Surg. 1980, 191, 511–512.
- Eyesan, S.U.; Obalum, D.; Abdulkareem, F.; Nnodu, O.; Idowu, O. Surgical consideration for benign bone tumors. Niger. J. Clin. Pract. 2011, 14, 146–150.
- Woertler, K. Benign bone tumors and tumor-like lesions: Value of cross-sectional imaging. Eur. Radiol. 2003, 13, 1820–1835.
- Hakim, D.N.; Pelly, T.; Kulendran, M.; Caris, J.A. Benign tumours of the bone: A review. J. Bone Oncol. 2015, 4, 37–41.
- Laliotis, N.; Chrysanthou, C.; Konstantinidis, P.; Papadopoulou, L. Osteoid Osteoma in Children Younger than 3 Years of Age. Case Rep. Orthop. 2019, 2019, 8201639.
- Gupta, S.; Sinha, S.; Narang, A.; Kanojia, R.K. Intramedullary osteoid osteoma in an 11-month-old child. J. Postgrad. Med. 2020, 66, 57–58.
- Frassica, F.J.; Waltrip, R.L.; Sponseller, P.D.; Ma, L.D.; McCarthy, E.F., Jr. Clinicopathologic features and treatment of osteoid osteoma and Osteoblastoma in children and adolescents. Orthop. Clin. N. Am. 1996, 27, 559–574.
- Pavone, V.; Testa, G.; Isfan, F.; Sessa, A.; Sessa, G.; Canavese, F. Metachronous osteoid osteoma of the mid diaphysis and proximal metaphysis of the tibia: Double localization at 5 years interval. World Cancer Res. J. 2018, 5, e1087.
- Atesok, K.I.; Alman, B.A.; Schemitsch, E.H.; Peyser, A.; Mankin, H. Osteoid osteoma and osteoblastoma. J. Am. Acad. Orthop. Surg. 2011, 19, 678–689.
- May, C.J.; Bixby, S.D.; Anderson, M.E.; Kim, Y.J.; Yen, Y.M.; Millis, M.B.; Heyworth, B.E. Osteoid Osteoma About the Hip in Children and Adolescents. J. Bone Jt. Surg. Am. 2019, 101, 486–493.
- Schulman, L.; Dorfman, H.D. Nerve fibers in osteoid osteoma. J. Bone Jt. Surg. Am. 1970, 52, 1351–1356.
- Tachdjian, M.O. Generalized affectations of the muscular skeletal system. In Clinical Pediatric Orthopedics: The Art of Diagnosis and Principles of Management; Appleton & Lange: Stamford, CT, USA, 1997; p. 369.
- Confavreux, C.B.; Borel, O.; Lee, F.; Vaz, G.; Guyard, M.; Fadat, C.; Carlier, M.C.; Chapurlat, R.; Karsenty, G. Osteoid osteoma is an osteocalcinoma affecting glucose metabolism. Osteoporos. Int. 2012, 23, 1645–1650.
- Dancer, J.Y.; Henry, S.P.; Bondaruk, J.; Lee, S.; Ayala, A.G.; de Crombrugghe, B.; Czerniak, B. Expression of masterregulatory genes controlling skeletal development in benign cartilage and bone forming tumors. Hum. Pathol. 2010, 41, 1788–1793.
- Kneisl, J.S.; Simon, M.A. Medical management compared with operative treatment for osteoid-osteoma. J. Bone Jt. Surg. Am. 1992, 74, 179–185.
- Gitelis, S.; Schajowicz, F. Osteoid osteoma and osteoblastoma. Orthop. Clin. N. Am. 1989, 20, 313–325.
- Orlowski, J.P.; Mercer, R.D. Osteoid osteoma in children and young adults. Pediatrics 1977, 59, 526–532.
- Alani, W.O.; Bartal, E. Osteoid osteoma of the femoral neck simulating an inflammatory synovitis. Clin. Orthop. Relat. Res. 1987, 223, 308–312.
- Iyer, R.S.; Chapman, T.; Chew, F.S. Pediatric bone imaging: Diagnostic imaging of osteoid osteoma. AJR Am. J. Roentgenol. 2012, 198, 1039–1052.
- Assoun, J.; Richardi, G.; Railhac, J.J.; Baunin, C.; Fajadet, P.; Giron, J.; Maquin, P.; Haddad, J.; Bonnevialle, P. Osteoid osteoma: MR imaging versus CT. Radiology 1994, 191, 217–223.
- Vigorita, V.J.; Ghelman, B. Localization of osteoid osteomas–Use of radionuclide scanning and autoimaging in identifying the nidus. Am. J. Clin. Pathol. 1983, 79, 223–225.
- Peyser, A.B.; Makley, J.T.; Callewart, C.C.; Brackett, B.; Carter, J.R.; Abdul-Karim, F.W. Osteoma of the long bones and the spine. A study of eleven patients and a review of the literature. J. Bone Jt. Surg. Am. 1996, 78, 1172–1180.
- Sharma, K.V.; Yarmolenko, P.S.; Celik, H.; Eranki, A.; Partanen, A.; Smitthimedhin, A.; Kim, A.; Oetgen, M.; Santos, D.; Patel, J.; et al. Comparison of Noninvasive High-Intensity Focused Ultrasound with Radiofrequency Ablation of Osteoid Osteoma. J. Pediatr. 2017, 190, 222–228.
- Arrigoni, F.; Napoli, A.; Bazzocchi, A.; Zugaro, L.; Scipione, R.; Bruno, F.; Palumbo, P.; Anzidei, M.; Mercatelli, D.; Gravina, G.L.; et al. Magnetic-resonance-guided focused ultrasound treatment of non-spinal osteoid osteoma in children: Multicentre experience. Pediatr. Radiol. 2019, 49, 1209–1216.
- Lucas, D.R. Osteoblastoma. Arch. Pathol. Lab. Med. 2010, 134, 1460–1466.
- Andrea, C.E.; Bridge, J.A.; Schiller, A. Osteoblastoma. In WHO Classification of Tumours of Soft Tissue and Bone, 4th ed.; Fletcher, C.D., Bridge, J.A., Hogendoorn, P.C., Mertens, F., Eds.; International Agency for Research on Cancer: Lyon, France, 2013; p. 279.
- McLeod, R.A.; Dahlin, D.C.; Beabout, J.W. The spectrum of Osteoblastoma. AJR Am. J. Roentgenol. 1976, 126, 321–325.
- Wan, Y.; Zhao, W.; Jiang, Y.; Liu, D.; Meng, G.; Cai, Y. β-catenin is a valuable marker for differential diagnosis of Osteoblastoma and osteosarcoma. Hum. Pathol. 2014, 45, 1459–1465.
- Fittall, M.W.; Mifsud, W.; Pillay, N.; Ye, H.; Strobl, A.C.; Verfaillie, A.; Demeulemeester, J.; Zhang, L.; Berisha, F.; Tarabichi, M.; et al. Recurrent rearrangements of FOS and FOSB define Osteoblastoma. Nat. Commun. 2018, 9, 2150.
- Copley, L.; Dormans, J.P. Benign pediatric bone tumors. Evaluation and treatment. Pediatr. Clin. N. Am. 1996, 43, 949–966.
- Nemoto, O.; Moser, R.P.; Van Dam, B.E.; Aoki, J.; Gilkey, F.W. Osteoblastoma of the spine. A review of 75 cases. Spine 1990, 15, 1272–1280.
- Theologis, T.; Ostlere, S.; Gibbons, C.L.; Athanasou, N.A. Toxic osteoblastoma of the scapula. Skelet. Radiol. 2007, 36, 253–257.
- Lucas, D.R.; Unni, K.K.; McLeod, R.A.; O’Connor, M.I.; Sim, F.H. Osteoblastoma: Clinicopathologic study of 306 cases. Hum. Pathol. 1994, 25, 117–134.
- Greenspan, A. Benign bone-forming lesions: Osteoma, osteoid osteoma, and Osteoblastoma. Clinical, imaging, pathologic, and differential considerations. Skelet. Radiol. 1993, 22, 485–500.
- Boriani, S.; Capanna, R.; Donati, D.; Levine, A.; Picci, P.; Savini, R. Osteoblastoma of the spine. Clin. Orthop. Relat. Res. 1992, 278, 37–45.
- Golant, A.; Lou, J.E.; Erol, B.; Gaynor, J.W.; Low, D.W.; Dormans, J.P. Pediatric osteoblastoma of the sternum: A new surgical technique for reconstruction after removal: Case report and review of the literature. J. Pediatr. Orthop. 2004, 24, 319–322.
- Berry, M.; Mankin, H.; Gebhardt, M.; Rosenberg, A.; Hornicek, F. Osteoblastoma: A 30-year study of 99 cases. J. Surg. Oncol. 2008, 98, 179–183.
- Guo, W.; Xu, W.; Huvos, A.G.; Healey, J.H.; Feng, C. Comparative frequency of bone sarcomas among different racial groups. Chin. Med. J. 1999, 112, 1101–1104.
- Larsson, S.E.; Lorentzon, R.; Boquist, L. Giant-cell tumor of bone. A demographic, clinical, and histopathological study of all cases recorded in the Swedish Cancer Registry for the years 1958 through 1968. J. Bone Jt. Surg. Am. 1975, 57, 167–173.
- Viswanathan, S.; Jambhekar, N.A. Metastatic giant cell tumor of bone: Are there associated factors and best treatment modalities? Clin. Orthop. Relat. Res. 2010, 468, 827–833.
- McDonald, D.J.; Sim, F.H.; McLeod, R.A.; Dahlin, D.C. Giant-cell tumor of bone. J. Bone Jt. Surg. Am. 1986, 68, 235–242.
- Chrcanovic, B.R.; Gomes, C.C.; Gomez, R.S. Central giant cell lesion of the jaws: An updated analysis of 2270 cases reported in the literature. J. Oral Pathol. Med. 2018, 47, 731–739.
- Montgomery, C.; Couch, C.; Emory, C.L.; Nicholas, R. Giant Cell Tumor of Bone: Review of Current Literature, Evaluation, and Treatment Options. J. Knee Surg. 2019, 32, 331–336.
- Papanastassiou, I.; Ioannou, M.; Papagelopoulos, P.J.; Arealis, G.; Mihas, C.; Iakovidou, I.; Demertzis, N. P53 expression as a prognostic marker in giant cell tumor of bone: A pilot study. Orthopedics 2010, 33, 307–312.
- Schwartz, H.S.; Juliao, S.F.; Sciadini, M.F.; Miller, L.K.; Butler, M.G. Telomerase activity and oncogenesis in giant cell tumor of bone. Cancer 1995, 75, 1094–1099.
- Smith, L.T.; Mayerson, J.; Nowak, N.J.; Suster, D.; Mohammed, N.; Long, S.; Auer, H.; Jones, S.; McKeegan, C.; Young, G.; et al. 20q11.1 amplification in giant-cell tumor of bone: Array CGH, FISH, and association with outcome. Genes Chromosomes Cancer 2006, 45, 957–966.
- Lau, Y.S.; Sabokbar, A.; Gibbons, C.L.; Giele, H.; Athanasou, N. Phenotypic and molecular studies of giant-cell tumors of bone and soft tissue. Hum. Pathol. 2005, 36, 945–954.
- Cowan, R.W.; Singh, G. Giant cell tumor of bone: A basic science perspective. Bone 2013, 52, 238–246.
- Campanacci, M.; Baldini, N.; Boriani, S.; Sudanese, A. Giant-cell tumor of bone. J. Bone Jt. Surg. Am. 1987, 69, 106–114.
- Murphey, M.D.; Nomikos, G.C.; Flemming, D.J.; Gannon, F.H.; Temple, H.T.; Kransdorf, M.J. From the archives of AFIP. Imaging of giant cell tumor and giant cell reparative granuloma of bone: Radiologic-pathologic correlation. Radiographics 2001, 21, 1283–1309.
- Aoki, J.; Tanikawa, H.; Ishii, K.; Seo, G.S.; Karakida, O.; Sone, S.; Ichikawa, T.; Kachi, K. MR findings indicative of hemosiderin in giant-cell tumor of bone: Frequency, cause, and diagnostic significance. Am. J. Roentgenol. 1996, 166, 145–148.
- Hamidi, S.; Mottard, S.; Berthiaume, M.J.; Doyon, J.; Bégin, M.J.; Bondaz, L. Brown tumor of the iliac crest initially misdiagnosed as a giant cell tumor of the bone. Endocrinol. Diabetes Metab. Case Rep. 2020, 2020.
- Gao, Z.-H.; Yin, J.-Q.; Xie, X.-B.; Zou, C.-Y.; Huang, G.; Wang, J.; Shen, J.-N. Local control of giant cell tumors of the long bone after aggressive curettage with and without bone cement. BMC Musculoskelet. Disord. 2014, 15, 330.
- Uglialoro, A.D.; Maceroli, M.; Beebe, K.S.; Benevenia, J.; Patterson, F.R. Distal femur defects reconstructed with polymethylmethacrylate and internal fixation devices: A biomechanical study. Orthopedics 2009, 32, 561.
- Cornelis, F.; Truchetet, M.; Amoretti, N.; Verdier, D.; Fournier, C.; Pillet, O.; Gille, O.; Hauger, O. Bisphosphonate therapy for unresectable symptomatic benign bone tumors: A long-term prospective study of tolerance and efficacy. Bone 2014, 58, 11–16.
- Savvidou, O.D.; Bolia, I.K.; Chloros, G.D.; Papanastasiou, J.; Koutsouradis, P.; Papagelopoulos, P.J. Denosumab: Current use in the treatment of primary bone tumors. Orthopedics 2017, 40, 204–210.
- Yang, Y.; Huang, Z.; Niu, X.; Xu, H.; Li, Y.; Liu, W. Clinical characteristics and risk factors analysis of lung metastasis of benign giant cell tumor of bone. J. Bone Oncol. 2017, 7, 23–28.
- Donthineni, R.; Boriani, L.; Ofluoglu, O.; Bandiera, S. Metastatic behaviour of giant cell tumour of the spine. Int. Orthop. 2009, 33, 497–501.
More