Historically, primary human progenitor cells (e.g., WI-38 and MRC-5 diploid-cell sources) have been industrially applied in research and in manufacturing processes for vaccines and for biologicals. Furthermore, tissue-specific primary progenitor-cell banks have recently been developed and exploited for the provision of safe, consistent, and effective cellular active pharmaceutical ingredients (API) in homologous allogeneic regenerative medicine applications. Notably, the modern legal and regulatory frameworks for novel therapeutic products and for progenitor-cell therapy development have been iteratively optimized to guarantee utmost product safety, quality, and efficacy. Over 50 years of global technical hindsight around progenitor-cell biotechnological substrates and over 30 years of in-house clinical experience around the therapeutic uses of standardized progenitor-cell sources in Switzerland have demonstrated the importance of such biological materials for public health. The aim of this entry work was to summarize the evolution of the industrial applications of selected primary progenitor-cell sources, ranging from the use as robust biotechnological substrates to standardized cellular API manufacture and their clinical uses in highly specialized regenerative medicine.
- biotechnological substrates
- cell therapies
- organ donation
- pharmacopeial monographs
- progen-itor cells
- quality requirements
- regenerative medicine
- regulatory compliance
- standardized transplants
- vaccine substrates
1.Introduction
Primary progenitor mammalian cells are characterized by extensive yet finite in vitro lifespans, defined tissue-specific phenotypes, and the technical potential for robust and extensive cell-batch-manufacture applications in controlled environments [1]. Primary progenitor cells are diploid cells, which are generally fibroblast-like in adherent in vitro monolayer cultures, and are non-modified, constituting cell types rather than cell lines [1,2]. Such cell sources were initially studied and were proposed as manufacturing substrates during the last third of the twentieth century, during the bold exploration of new frontiers in biotechnology for novel therapeutic-product development [3–5]. Specifically, the urgent global need for standardized substrates in the field of industrial vaccine-product manufacture had prompted the search for optimal and safe cell sources [6]. Therefore, notorious applied cellular-biology studies from the 1960s have laid the foundations of most modern biotechnology processes, with the original establishment and the subsequent thorough characterization of well-known diploid cell types (e.g., WI-38 and MRC-5 fetal lung fibroblast sources) [3,5]. Such specific biological materials, stabilized in cryopreserved form and in defined cell-bank systems, were soon proposed, confirmed, and were industrially adopted as technically optimal and high-quality biotechnological substrates. Thereafter, diversified and extensive industrial experience was gathered around these original diploid-cell sources, along with many demonstrated and tangible gains for global public health [7,8]. Notably, wide arrays of vaccine products were developed and/or produced using the WI-38 or the MRC-5 diploid cells, directly contributing to the effective prevention of, among other human diseases and affections, chickenpox, hepatitis A, poliomyelitis, smallpox, rabies, and rubella [6,7].
Interestingly, the direct and indirect use of such primary progenitor cell sources by scientific researchers and by pharmaceutical industries has constituted the basis for continued ethical and moral debates [9–16]. Despite the documentation of many proven public health benefits of using diploid cells for life-saving therapeutic-product development and manufacture, thorough discussions have been driven notably by religious scholars around the context of the original tissue procurement [7,14–16]. Nonetheless, the intensive industrial use and the global material demand for high-quality biotechnological substrates have currently never been higher, prompting the development of novel diploid-cell sources and the renewal of aging cell stocks [17–19]. Therefore, several sustainability and stability characteristics of appropriately established primary progenitor-cell sources are being set forth as critical attributes and as major technical advantages [1,17]. Based on such quality-oriented considerations, the development and the qualification of novel standardized progenitor-cellular substrates are of high current interest, for eventual valorization in the supply chain of modern biotechnological industries [6].
In parallel to the industrial manufacturing applications, where primary progenitor cells are used as ancillary biotechnological substrates, high interest has been recently set on the direct use of the same types of cells as starting materials and as raw materials in cytotherapeutic products (Figure 1) [1,2]. Indeed, several technical and biological characteristics of such tissue-specific cell sources confer tangible advantages for the therapeutic uses thereof as active pharmaceutical ingredients (API) in homologous allogeneic regenerative-medicine applications [6,20]. Therefore, it has been reported that, when appropriately sourced and bioprocessed, primary progenitor-cellular APIs may be considered as optimally adapted for industrial transposition and for clinical translation in modern tissue-engineering applications [20]. In addition to the documented vast therapeutic potential and low risks of immunogenicity, the scalability and the robustness of selected primary progenitor-cell sources enable the eventual use of safe and consistent cytotherapeutic APIs [20–27]. Importantly, the use of stringent methodological workflows for progenitor-cell sourcing and for the subsequent clinical applications currently appear as central in the overall therapeutic approach, with specified ownership, rights, and obligations related to the defined cell sources [2,28,29].

Figure 1. Schematic technical overview of the established workflows for optimized primary progenitor-cell-type establishment and of related industrial applications. (A) Using ad hoc and well-defined methodological processes for the original tissue procurement, the appropriate in vitro primary-cell-isolation procedures are applied to obtain a homogenous preliminary pool of primary progenitor cells. (B) Following stringent manufacture-optimization steps, the multi-tiered GMP cell banking is performed to constitute the primary progenitor-cell banks. (C) The established and qualified cell stocks of primary progenitor cells may then be used as biotechnological substrates or as cytotherapeutic APIs in specialized regenerative medicine. API, active pharmaceutical ingredient; DMEM, Dulbecco’s modified Eagle medium; FBS, fetal bovine serum; GMP, good manufacturing practices; MCB, master cell bank; QC, quality control; WCB, working cell bank.
Despite several recent regulatory hurdles and bottlenecks affecting the global development of many cell-based therapeutic products, numerous efforts and vast resources have been allocated toward preclinical and clinical work around the therapeutic use of primary progenitor cells [30–37]. Specifically, almost three decades of specialized clinical experience with such progenitor-cell-based tissue-engineering products (TEP) have been gathered in pediatric burn-patient care [38–41]. Polyvalent use of skin-derived diploid progenitor fibroblasts as viable cellular APIs indicated for the promotion of cutaneous wound healing has revealed unique capacities for the obtention of structural and of functional restoration of the affected cutaneous structures [38,41]. In particular, the continued therapeutic management of pediatric burns and of chronic inflammatory cutaneous wounds in the Lausanne University Hospital (Lausanne, Switzerland) using local homologous progenitor cell therapies since the 1990s may be considered as a landmark in the field [38,42,43].
Furthermore, the continued work in applied bioengineering and on diversified tissue-specific progenitor cell-therapeutic applications has revealed similar high potential for the allogeneic treatment of soft-tissue and of musculoskeletal-tissue affections [44–50]. Therefore, using evolutive process-based and conserved methodological aspects of diploid progenitor-cell sourcing, multi-tiered cell-bank establishment, and cellular API processing, it was shown that qualified progenitor cell sources were well-adapted for the establishment of safe and sustainable therapeutic-material supply chains (Figure 1) [1,51–53]. Therefore, it can be summarily stated overall that selected primary progenitor-cell sources constitute scientifically and historically proven robust and polyvalent tools, to be indirectly or directly applied for the manufacture of therapeutic products, thereby tangibly contributing to the global betterment of public-health capitals [1].
2. Methodological Aspects of Starting Biological Material Procurement for Modern Primary Progenitor-Cell-Type Establishment
The drastic evolution in medical laws, in ethical guidelines, and in therapeutic practices differentiates the context of establishment of the original diploid-cell sources in the 1960s and of current research and development involving novel primary progenitor-cell sources [3,6]. Specifically, the lack of harmonized methodology or of documented verification of consent for the donors of the HeLa or the WI-38 cell sources have been the object of public scrutiny and the origin of major debates over the past half-century [29]. Therefore, the recent methodological and regulatory frameworks for tissue and cell procurement in view of clinical applications were iteratively optimized. This was specifically performed in order to notably guarantee minimal ethical footprints of the considered cytotherapeutic API sources, as well as the utmost safety and quality of the progeny materials (e.g., cell-based or cell-derived treatments) [2,28]. In Europe, the requirements for tissue/cell-donor identification, qualification, and inclusion in a research or a development program are based notably on Directives 2006/17/EC and 2006/86/EC of the European Commission and on Directive 2004/23/EC of the European Parliament and Council, which are referenced in the “Guide to the quality and safety of tissues and cells for human application” of the European Directorate for the Quality of Medicines and Healthcare (EDQM) [54–57]. In Switzerland, the same research must conform to federal laws on transplantation and on therapeutic products [58,59]. Additionally, the proper ethical validation must be sought locally for specific research programs [6,28].
In order to conform to all the traceability and the quality requirements of biological material sourcing for eventual therapeutic applications, the procurement of specific tissues for progenitor-cell sourcing was notably organized under the Swiss progenitor-cell-transplantation program since the 1990s [2,28]. Originally registered with the Swiss federal office of public health (FOPH), this cell-transplantation program was iteratively adapted and was updated in accordance with the requirements of Swissmedic, the Swiss institute for therapeutic products [1,2]. Importantly, it is to be noted that Swiss law allows women to voluntarily and legally interrupt a pregnancy up until twelve weeks post-amenorrhea, following which an organ donation may be made [2]. Furthermore, tissues or cells may be isolated from a pre-natal organ donation and may be used only in the case where documented donor consent has been provided, and such consent may be sought only after the decision for the gestation interruption has been confirmed [59]. Therefore, exhaustive information must be provided and must be confirmed in writing to potential tissue donors regarding the planned and the potential uses of the donated materials and of derived progeny materials, including all commercial applications [1,2,28]. In addition, donor consent must also be documented for the extensive testing to be performed on the donor and on the donated materials, for the insurance of maximal safety and optimal biological quality [20]. A defined donor reconsideration period is provided, for appropriate reflection and confirmation of understanding and for consent provision [28]. Importantly, the appropriate compartmentalization and a complete independence must be insured between the medical staff handling tissue donors and the staff performing the bioprocessing of the donated materials [28]. This aspect has been integrated within the definition of the organigram for the Swiss progenitor-cell-transplantation program, aiming for an optimal exploitation of the available multidisciplinary professional competences (Table 1, Figure S1).
Table 1. Multidisciplinary personnel involved in the different compartments of a progenitor-cell-transplantation program. The existence of defined roles and responsibilities that are specified for each stakeholder enables the optimal compliance with applicable legal requirements regarding necessary restrictions in the information and material flows. GMP, good manufacturing practices; QC, quality control.
|
Personnel Identification |
Key Professional Competences |
Roles in the Cell-Transplantation Program |
|
Program Manager 1 |
Experience in tissue and cell banking. Qualifications for GMP cell manufacture. Experience in multidisciplinary professional group coordination. |
Selection of the optimal biological starting materials. Establishment and coordination of the cell-transplantation program; acting as the legally responsible person. Custodian of the cell-transplantation-program records and biobank administrator. |
|
Technical Manager 1 |
Experience in tissue and cell banking. Qualifications for GMP cell manufacture. |
Selection of the optimal biological starting materials. Oversight of the tissue processing, in vitro cell culture initiation, and of cell banking. Responsible person for GMP processes. |
|
Medical Doctor |
Experience in medical pregnancy termination. |
Screening of potential donors, eligibility documentation, procurement of the donation, and implementation of the biospecimen and information coding. |
|
Legal Advisor |
Experience in applicable laws and regulations on transplantation and on therapeutic products. |
Support in the establishment of the cell-transplantation program and liaison with local ethics committees and with local or national health authorities. |
|
Pathologist |
Experience in pre-natal pathology and in histopathology. |
Anatomical and histopathological QC examination of the donated tissues. |
|
Immunologist |
Experience in medical analytics. Qualifications and accreditations for pathogen screening and sample analysis. |
Screening for pathogen detection in the blood samples of potential donors and QC testing of the donated materials and of the progeny cells. |
1 Role generally assumed by an experienced biologist, a pharmacist, or a bioengineer.
Appropriate documentation of the specific donor consent and full insurance of donor anonymity preservation are of critical methodological importance within a progenitor-cell-transplantation program. Furthermore, building on the historical experience gained around biobank administration for the WI-38 or the HeLa cell sources, highest attention should be paid to the precise definition of rights and obligations with regard to the ownership of specific cells’ sources and of the related technical know-how [1,29]. Technically, the implementation of the stringent quality-related procedures and workflows for tissue sourcing and for the subsequent cell-culture steps are pre-requisites for the eventual use of primary cells as biotechnological substrates or as therapeutic materials [57]. Based on ad hoc risk analyses, specific biosafety-testing schemes are established, specifying sequential serological screening of the donor before the donation and after a reconsideration period, for the exclusion of sero-conversion for specified pathogens [28]. Preliminary qualification of the potential donors and follow-up testing are performed by a medical doctor, based on the following minimal and cumulative inclusion criteria:
- Donor of female sex and female gender.
- Donor of biological age between 18 years and 25 years.
- Donation performed at 12–14 weeks following conception.
- Donor of specified nationality and place of residence.
- Donor pregnancy resulting from natural insemination.
- Donor performing voluntary, gratuitous, and legal donation.
- Documentation of informed consent for the gestation termination.
- Documentation of informed consent for the donation.
- Donor in good overall health.
- Donor not suffering from any chronic disease or affection.
- Qualifying serological screening of the donor for specified pathogens (e.g., HIV, HBV, HCV, HTLV, CMV, S-West Nile virus, Treponema pallidum, Toxoplasma gondii, etc.).
- Availability of the donor after three months for repeat serological testing.
- No treatment of the donor with anti-inflammatory or other therapeutic drugs in the past six months.
- No inoculation of the donor with vaccines in the past four weeks.
- No donor history of post-travel sickness in the past six months.
- No history of immunotherapeutic treatment of the donor.
- No history of transplantation receipt by the donor.
- No history of Creutzfeldt–Jacob disease in the donor’s family.
Following the qualification and inclusion of the donor in the progenitor-cell-transplantation program, specific organs, tissues, and related anonymized medical information are provided for the appropriate testing and for further in vitro bioprocessing, in view of primary progenitor-cell-culture initiation. Importantly, it is to note that all the donated materials and the derived progeny cell sources must be quarantined until the donor has been screened after the specified reconsideration and follow-up period, at which point the inclusion in transplantation program must be confirmed by the donor [2]. In case of out-of-specification testing results from the biosafety screen or in case of retraction of the donor with regard to the inclusion in the cell transplantation program, all the materials and the cryopreserved cell stocks are necessarily destroyed [2]. Concerning the ethical and legal oversight of the cell-transplantation program, appropriate controls must be implemented, and rules must be defined in the approved framework documents (e.g., biobank regulations) [28].
Important methodological evolution around the organ-donation traceability and quality aspects since the initial establishment of diploid-progenitor-cell sources in the 1960s has demonstrated the need for careful planning and adaptation of the processes to applicable legal frameworks [6]. Indeed, the exhaustive documentation of all the information related to cell sourcing and to the initial manufacturing steps constitutes the basis of a cell-type master file, which is to be referenced in all the processes or applications in which the specific progenitor-cell source is subsequently used [51]. Therefore, the constitution of specific cell-transplantation programs and of dedicated progenitor-cell biobanks within the appropriate quality-assurance systems has been proven to guarantee the optimal material and information traceability for the potential development of commercial substrates or of therapeutic products with derivatives of the donated biological material [1].
3. Optimized Modern Industrial Manufacturing of Primary Progenitor Cells in Tiered Cell-Bank Systems
The original interest and the early applications of primary diploid-cell sources within industrial manufacturing settings revolved mainly around vaccine development and production efforts during the twentieth century [6]. Therefore, as the replication of viral materials depended on the availability of the appropriate biological substrates in considerable quantities, tissue explants and pre-natal lung-derived fibroblasts were successively used for the production of diverse vaccines [1,6,7]. However, major safety concerns rapidly led to the instauration of stringent production-quality requirements for cell-based biotechnological substrates, notably including risk-based analyses and adaptable biosafety-testing schemes (Table 2) [6,60–65].
Table 2. Example of a biosafety-testing scheme and of cellular substrate qualification requirements applicable for cell substrates used in the GMP production of vaccines for human use, based on the Ph. Eur. 9.0 general chapter 5.2.3., 01/2017:50203 [65]. Required testing is indicated by a “+” symbol. Non-required testing is indicated by a “−” symbol. ECB, extended cell bank; EOPCB, end-of-production cell bank; GMP, good manufacturing practices; MCB, master cell bank; PCB, parental cell bank; Ph. Eur., European pharmacopoeia; WCB, working cell bank.
|
Material Safety and Quality Assessments: Testing Class and Testing Parameters |
Considered Cell Bank Tiers and Applicability of Testing |
||||
|
Cell Seed/PCB |
MCB |
WCB |
ECB/EOPCB |
||
|
1. Identity and Purity |
Cellular morphology |
+ |
+ |
+ |
+ |
|
Cell-type identification (genetic and biochemical, immunological, or cytogenetic profiling) |
+ |
+ |
+ |
+ |
|
|
Cell-type karyotype (diploid cells) |
+ |
+ |
+ (1) |
+ (1) |
|
|
Cell type in vitro lifespan (diploid cells) |
− |
+ |
+ |
− |
|
|
2. Extraneous Agents |
Tests for bacterial and fungal contamination |
− |
+ |
+ |
− |
|
Tests for mycobacteria |
− |
+ (2) |
+ (2) |
− |
|
|
Tests for mycoplasmas |
− |
+ |
+ |
− |
|
|
Tests for spiroplasmas (3) |
− |
+ |
+ |
− |
|
|
Electron microscopy |
− |
+ (4) |
− |
+ (4) |
|
|
Tests for extraneous agents in cell cultures (intact cells or equivalent cell lysates) |
− |
+ |
+ |
+ |
|
|
Tests in animals and in eggs |
− |
− |
+ (5) |
+ (5) |
|
|
Tests for specific viruses |
− |
+ (6) |
+ (6) |
+ (6) |
|
|
Tests for retroviruses |
− |
+ (4) |
− |
+ (4) |
|
|
3. Tumorigenicity |
Cell-type tumorigenicity |
+ (7,8) |
− |
− |
+ (7) |
(1) Diploid cellular characteristics were determined for each WCB by using cells at a population-doubling level ≥ to that of cells used for production batches. (2) In case of cell-type susceptibility to infection by Mycobacterium tuberculosis or by other Mycobacterium spp. (3) In case insect cells or plant-based raw materials are used. (4) Determined for the MCB by using cells at a population-doubling level ≥ to that of cells used for production batches. (5) Determined for each WCB by using cells at a population-doubling level ≥ to that of cells used for production batches. (6) Specific search for potential contaminants based on a risk analysis taking into account the origin of the cells, potential adverse introduction of an extraneous agent during cell manufacture, and use of animal-derived raw materials. The appropriate testing stage was selected based on the risk analysis. (7) Cell types and cell lines recognized as being non-tumorigenic (e.g., MRC-5, WI-38, and FRhL-2) or cell types and cell lines known or presumed to be tumorigenic (e.g., CHO and BHK-21) were not tested. (8) Determined for the cell seed/PCB by using cells at a population doubling level ≥ to that of cells used for production batches.
Therein, stringent quality-oriented procedures and highly controlled access to materials and to information were implemented for the tissue procurement and for further bioprocessing, to ensure optimal safety and quality of the progeny cellular materials [28]. For the original cell isolation and initiation of the in vitro culture of the primary progenitor cells from the donated tissues, an enzymatic- or a mechanical-processing workflow may be used (Figures S2–S4) [1]. Notably, a standard cell-proliferation medium (e.g., DMEM supplemented with fetal bovine serum) and simple in vitro culture conditions (e.g., 5% CO2 and 37 °C incubation) are sufficient for the rapid initiation of the adherent monolayer primary progenitor-cell cultures in GMP settings (Figures 2 and S5) [6].

Figure 2. Illustrative overview of the main technical phases and steps required for the culture initiation of primary progenitor cells and for cell stabilization in a tiered cell-bank system. (A) Tissue procurement following the organ donation. (B) Mechanical tissue treatment in the primary culture vessel. (C) Proliferation medium dispensing for the promotion of preliminary cell growth. (D) Primary in vitro cell outgrowth in adherent monolayer. Scale bar = 75 µm. (E) Proliferation medium-exchange step. (F) Incubation of cell cultures. (G) Verification of cell monolayer confluency by contrast-phase microscopy. (H) Cell-culture harvest by trypsinization. (I) Cell-count determination by hemocytometer enumeration. (J) Cell-suspension conditioning in individual vials for cell cryopreservation. (K) Loading of cell vials in freezing devices for constant-rate cooling. (L) Long-term cryostorage of cell vials in the ad hoc progenitor-cell biobank.
Based mainly on the high robustness of in vitro processing and on the extensive inherent proliferative potential of primary progenitor cells, such biological materials may be handled under good-manufacturing-practice (GMP) requirements and may be used for serial cell banking at industrial scales [20,66]. Following the pre-defined technical specifications and biobanking strategies, the cell seed lots or the preliminary cell pools may be used as cellular starting materials for the sequential establishment of multi-tiered progenitor cell stocks (i.e., cryopreserved vial lots with 106 to 107 cells/vial) (Figures S6 and S7). Following the appropriate quality-control testing, the various cell bank lots may be released from production, with the cell-bank-tier nomenclature being defined by the respective in vitro passage levels or the cell-population-doubling levels of the considered cells [66]. Therefore, the cell batches or lots may be classified using the specific nomenclature, depending on the in vitro cell age, as belonging to the preliminary cell pool or cell seed, to the parental cell bank (PCB), to a master cell bank (MCB), or to a working cell bank (WCB). Additionally, extended cell banks (ECB) or end-of-production cell banks (EOPCB) are generated, for the appropriate quality and safety testing of the cell type of interest (Tables 2 and 3) [20,65,67].
Table 3. Nomenclature definition in relation to multi-tiered cell banking of primary progenitor cells in biotechnology, as set forth in Ph. Eur. general chapters 5.2.1. and 5.2.3. and/or under the Swiss progenitor-cell-transplantation program [1,65,67]. API, active pharmaceutical ingredient; ECB, extended cell bank; EOPCB, end-of-production cell bank; MCB, master cell bank; PCB, parental cell bank; Ph. Eur., European pharmacopoeia; WCB, working cell bank.
|
Nomenclature Terms |
Term Definitions/Material Descriptions |
|
Cell Bank System |
A system whereby the successive final lots of a product are manufactured by culture in cells derived from the same MCB 1. A number of containers from the MCB are used to prepare a WCB. The in vitro age of the cells is counted from the MCB 2. The cell-bank system is validated for the highest in vitro passage level achieved during routine production 3. The use, identity, and inventory control of the individual cell-bank containers is carefully documented. |
|
Diploid Cell Type |
Cryopreserved stocks and cultures of diploid cells that have a high but finite capacity for multiplication in vitro 4. In diploid cell types, the cells have essentially the same characteristics as those of the tissues of origin. |
|
Primary Cell Cultures |
Cultures of cells directly obtained by trypsinization or by mechanical treatment of a suitable starting tissue or organ fragment 5. The cells are essentially identical to those of the tissue of origin and are no more than five in vitro passages from the initial preparation from the mammalian tissue of origin. The primary cell cultures are harvested to form the preliminary cell pool. |
|
Parental Cell Bank (PCB) |
A preliminary cell pool distributed into containers in a single operation, processed together, and stored in such a manner as to ensure uniformity and stability and to prevent contamination 6. |
|
Master Cell Bank (MCB) |
A culture of cells derived from the cell seed/PCB, distributed into containers in a single operation, processed together, and stored in such a manner as to ensure uniformity and stability and to prevent contamination. The MCBs are usually stored at −70 °C or at lower temperatures 7. |
|
Working Cell Bank (WCB) |
A culture of cells derived from a MCB and intended for use in the preparation of production cell cultures. The WCB lot is distributed into containers, processed, and is stored as described for the MCBs. |
|
Production Cell Culture |
A culture of cells intended for use in production; it may be derived from one or more containers of a WCB or it may be a primary cell culture. |
|
End-of-Production Cell Bank (EOPCB) or Extended Cell Bank (ECB) |
A culture of cells derived from a WCB, at or beyond the maximum in vitro cell-population-doubling level used for production, distributed into containers in a single operation, processed together, and stored in such a manner as to ensure uniformity and stability and to prevent contamination 3. |
|
Final Lot or Batch |
A collection of closed, final containers or other final dosage units that are expected to be homogenous and equivalent with respect to risk of contamination during filling or preparation of the final product. The dosage units are filled, or otherwise prepared, from the same final bulk and are closed in one continuous working session. They bare a distinctive number or code identifying the final lot or batch. |
1 This statement is valid for the use of cells as biotechnological substrates, as the cells themselves are the intermediary product or the API in regenerative medicine. 2 The in vitro cell age is counted from the cell isolation step in the Swiss progenitor-cell-transplantation program. 3 The cell-bank system is validated using an ECB/EOPCB at an in vitro passage level 50% above the API production level in the Swiss progenitor-cell-transplantation program. 4 Immortal cells constitute a cell line, whereas non-modified primary diploid cells generally constitute a cell type. 5 Primary progenitor-cell cultures may be obtained by enzymatic or by mechanical tissue treatment in the Swiss progenitor-cell-transplantation program. 6 The PCB tier is not defined in the Ph. Eur., which describes a cell seed. 7 For optimal stability, primary progenitor-cell-bank vials should be stored in the gaseous or in the liquid phase of liquid nitrogen in dedicated storage tanks.
The use of simple and standard in vitro cell culture materials, reagents, and conditions is conserved throughout the serial expansion phases of the primary progenitor-cell-banking campaigns. However, a crucial technical-optimization phase should be carried out on a pilot progenitor-cell bank following each new primary cell-type establishment and qualification procedure, for optimization of the manufacturing yields and of the overall biological material quality [51]. Specifically, close attention must be paid to benchmarking of the cell-culture vessels (i.e., the vessel type, the model, the culture surface, and the gas-management method), the cell-growth-medium supplements (i.e., the supplier and lot number), and of the culture-handling workflows (i.e., the cell-culture-medium volumes, the medium-exchange rates, the cell-seeding densities, and the cell-harvesting timepoints) (Figure S7) [6,51]. Then, once the cell type-specific optimal parameters and specifications have been established and validated, the industrial GMP progenitor-cell-banking campaigns may be initiated. Following quality-assurance (QA) requirements, appropriate in-process controls (IPC) and post-process controls (PPC) should be implemented during cell manufacturing, in relation to the specified targets and acceptance criteria [51]. Depending on the tier of the individual cell-production lots and of the corresponding cell banks, the appropriate product characterization and release testing are performed (e.g., recovery assays, isoenzyme testing and DNA fingerprinting, sterility testing, or research of specified and of unspecified microorganisms with a particular focus set on viruses of human, bovine, and of porcine origin) (Table 2) [20,66]. Furthermore, complementary and specific testing is performed on ECB/EOPCB materials (e.g., karyology studies and in vitro and in vivo tumorigenicity assays) for the qualification of safety, quality, and stability of the considered primary progenitor-cell sources (Table 2) [1,51,66].
Overall, due to the favorable inherent technical specificities existing for primary progenitor cells, such as the rapid establishment and the extensive cell-banking capabilities, the necessity for repeated organ donations is virtually negated. This aspect is tangibly confirmed by the historical industrial use of the WI-38 and MRC-5 diploid-cell sources for example, which have lasted over fifty years to date [7,19]. The use of such stable and characterized biological standards contributes to augment the process and patient safety and to lower the direct manufacturing costs, due to the extensive documented hindsight available for the considered cell substrates. The reported robustness of selected primary progenitor-cell sources may be notably attributed to conservative cell-isolation processes, extensive inherent expansion capacities, minimal growth requirements in culture, and to high resistance to oxidative stress [1,2]. Based on such attributes, the multi-tiered primary progenitor-cell biobanks may be rapidly and easily established and may be qualified under current GMP (cGMP) standards using the technically optimized manufacturing frameworks and workflows [51]. Despite the finite in vitro lifespan of primary progenitor cells and the existing restrictions of use at higher in vitro passage levels, it was mathematically established that a single organ donation was potentially sufficient for the standardized derivation of several billion progenitor-cell WCB vials [20]. Such technical possibilities have paved the way toward the sustainable exploitation of homogenous biotechnological substrate materials over extended periods, in particular during times of acute industrial demand for vaccine-substrate materials (e.g., the current COVID-19 pandemic) [6].
4. Industrial Applications of Primary Progenitor Cells as Biotechnological Substrates or as Therapeutic APIs and Related Quality Requirements
As previously mentioned, specific diploid-cell sources (e.g., WI-38 cells) have been recognized as instrumental in reducing mortality and in empowering the protection of public-health capitals during the twentieth century [7,16]. This was notably attained by the judicious application of primary pre-natal lung-tissue-derived cell types as biotechnological substrates in the vaccine industry, which requires specific cell-culture steps for product development and for manufacturing activities (Table 4) [6].
Table 4. Notable vaccine product types for which the industrial manufacturers are known to have used the WI-38 1 and the MRC-5 2 diploid-cell sources in the development and production activities [2]. ATCC, American-type culture collection; MRC, medical research council; WI, Wistar institute.
|
Target Diseases |
Diploid Cell Substrate Types |
Identified Industrial Manufacturers |
|
Smallpox |
MRC-5 |
Acambis |
|
Chickenpox |
WI-38, MRC-5 |
Merck, GlaxoSmithKline |
|
Shingles |
WI-38, MRC-5 |
Merck |
|
Poliomyelitis |
MRC-5 |
Sanofi Pasteur |
|
Mumps |
WI-38 |
Merck |
|
Measles |
WI-38 |
Merck, GlaxoSmithKline |
|
Rubella |
WI-38 |
Merck |
|
Hepatitis A |
MRC-5 |
Merck, GlaxoSmithKline, Sanofi, Berna |
|
Hepatitis B |
MRC-5 |
GlaxoSmithKline, Sanofi |
|
Typhoid |
MRC-5 |
Sanofi Pasteur |
|
Rabies |
MRC-5 |
Sanofi Pasteur |
1 The WI-38 cell type (i.e., Wistar Institute 38) is currently commercially available from the ATCC under the brand name ATCC® CCL-75™ (https://www.atcc.org/products/ccl-75, accessed on 1 December 2021). 2 The MRC-5 cell type (i.e., Medical Research Council 5) is currently commercially available from the ATCC under the brand name ATCC® CCL-171™ (https://www.atcc.org/products/ccl-171, accessed on 1 December 2021).
The most documented and historically used diploid-cell sources are the WI-38 and the MRC-5 cell types, isolated in the 1960s in the USA (Hayflick et al.) and in the UK (Jacobs et al.), respectively [3–5]. Initially studied mainly as models of cellular aging and as tools of developmental cellular biology, such stabilized cellular materials were soon proposed, swiftly adopted, and extensively qualified as highly efficient biotechnological cell substrates [2]. The most notable original optimization and thorough characterization work of Dr. Leonard Hayflick had indeed revealed the extensive expansion potential, stability, and safety of selected primary diploid cell types (e.g., WI-38, a pre-natal lung tissue-derived diploid cell source) [3,4,7]. These elements, along with several intrinsic technical aspects of primary diploid-cell sources, have been set forth as the critical and key attributes of ideal vaccine substrates in the 1960s. Such attributes have been summarized by the World Health Organization (WHO) in several reference documents, notably due to the key implications of vaccines in public health [6,18,19]. Several of these elements, pertaining to the technical rationale and to the characterization of reference cell banks, may contribute to shed some light on the value (i.e., technical aspects and for public health) or on the historical extensive use of such cell substrates and are summarized as follows:
- Full traceability may be made available back to the (anonymized) origin of the primary cell source, around the derivation of the primary cell type and around the materials used in the preparation of the cell seed stocks.
- The technical possibility exists to create, by means of serial in vitro sub-cultures in a defined sustainable cell-bank system, sufficient amounts of consistent populations of cells to satisfy a globalized industrial-scale demand for high-quality biological substrates (e.g., millions of standardized WCB vials of the same original cell type).
- The technical possibility exists to cryopreserve extensive and homogenous cell lots at relatively early population-doubling levels or passage levels within the qualified in vitro lifespan of the primary cell type of interest.
- A standardized approach may be adopted for the original establishment of appropriate cell types and for the characterization of the derived cryopreserved cell banks, which may later be used as starting materials for further multi-tiered cell banking and for the sustainable provision of standardized cell sources for research and industrial applications.
- The technical possibility exists (i.e., due to the high sustainability of the cell sources) for the implementation of extensive and appropriate (i.e., risk analysis-based) biosafety testing schemes for the qualification of manufacturer-specific cell banks before the use in vaccine-production activities.
- Numerous technical possibilities exist for the demonstration that the considered cellular materials are exempt from detectable adventitious agents and that they are unable to form tumors when inoculated into immunosuppressed animal models or equivalent models.
- The possibility exists that the original research is subject to open international scientific scrutiny and to collaborative technical investigations of the characteristics of the cells or of the possible presence of adventitious agents. The cell-characterization results may be peer-reviewed and published.
- A single source of cells may exist, with a growing scientifically and technically updated body of safety-testing data and a safe history of use, giving increased confidence to manufacturers, regulators, and public-policy makers.
- The supply of cells may be free of any constraint related to intellectual-property rights on final products.
- The possibility exists, based on intrinsic cellular characteristics, to propagate diverse viral materials (i.e., viral pathogens infecting humans) with extremely high efficiency of replication in the manufactured primary cell populations.
Of major technical importance, it should be noted that these human diploid-cell sources originally established in the 1960s remain in industrial use to this day, attesting to the highly sustainable nature of the appropriate exploitation of specific cell-bank systems. Particularly, non-transformed diploid cell types constitute substrates of choice for efficient viral-material propagation (i.e., the use of the cells as biological substrates to produce viral materials, which need cellular hosts for replication), as they may be considered as universal virus carriers (i.e., including SARS viruses) [6,17]. With regard to the technical aspects and the current uses of diploid cells by vaccine manufacturers, distinctions can be made as regards the process in which the cell substrates are used, which may comprise the following:
- (i) Product and process development stages, in order to identify the optimal mechanisms for viral-material replication and to select the optimal manufacturing processes and materials to be used for production.
- (ii) Process confirmation or validation stages, in order to ensure that the selected processes and materials may be tangibly transposed into production.
- (iii) Production-validation stages, in order to validate that the selected substrates, materials, and processes may be tangibly used in the final manufacturing system for the final-product formula.
Despite the aforementioned high-sustainability parameters of the original diploid-cell sources, the recent exponential increase in the global material demands and the intensive research for the development of novel vaccines since 2019 (e.g., against SARS-CoV-2) have outlined several bottlenecks in the global supply chain of vaccine substrates [6]. Indeed, the conjunction of the transiently elevated industrial activity and of the natural aging or instability of the original cell stocks has had a material depleting effect on most high-quality cell sources [17,19,68]. Indeed, despite the intense manufacturing workflow optimization driven by leading biotechnological industries, insufficient efforts and resources have been allocated toward the original re-establishment of novel robust primary cell types in view of substrate homologation [6]. As previously mentioned, many vaccine products (e.g., rubella, chicken pox, hepatitis A vaccines, etc.) have been developed and/or manufactured using processes including such diploid-cell types, despite the under-documented aspects of the original tissue procurement and of donor consent (Table 4) [6,69,70]. This aspect, along with the specified need for insurance of the utmost material safety and quality, have led to the progressive implementation of many industrial guidelines and requirements aiming to establish best practices, which essentially focus on multifactorial risk reduction [71–85].
Furthermore, based on the existing frameworks and on the extensive historical industrial experience with diploid cellular substrates, recent developments of bioengineering and of regenerative medicine have led to the investigation of the direct use of such cells as therapeutic APIs [1,2,6,20]. Therefore, the industrial applications of modern primary progenitor-cell sources may be considered as twofold, following identical biotechnological manufacturing processes, with the eventual exploitation as high-quality substrates or as therapeutic starting materials or as raw materials (Figure 3) [6].

Figure 3. Schematic technical overview of the evolution of primary progenitor-cell applications since the 1960s. In addition to the continued use as renewed biotechnological substrates (A), modern frameworks consider clinical-grade primary progenitor cells as APIs in homologous allogeneic regenerative medicine (B). The provided illustrated example is based on the historical biotechnological use of the WI-38 and the MRC-5 cell sources by the vaccine industry and on the current considerations for dual valorization options for modern lung-tissue-derived primary progenitor-cell sources (e.g., FE002-Lu cell type). API, active pharmaceutical ingredient; CH, Helvetic Confederation; PRC, People’s Republic of China; UK, United Kingdom; USA, United States of America.
Therefore, specific and additional quality requirements have been implemented, due to the use of primary progenitor cells or of cell derivatives as starting materials and as raw materials in therapeutic products, instead of as ancillary materials during biotechnological production [86–90]. Based on the aforementioned technical and biological characteristics of primary progenitor-cell sources, and due to the low inherent risks of immunogenicity or of tumorigenicity after cell transplantation, major clinical gains may result from the diverse therapeutic uses of these biological APIs [20,21,28]. Furthermore, due to the potential to derive extensive and consistent cell banks and homogenous final-product lots, primary progenitor cells bare the technical potential to constitute standard or quasi-universal components of allogeneic therapeutic products [1,20].
Importantly, the extensive in vitro expansion potential of selected primary progenitor-cell types enables the implementation of thorough biosafety-testing schemes during the API manufacturing steps, as well as cost-effective product preparation [20]. High importance is set on the safety of transplantation materials in human medicine, where robust primary cell sources may be fully characterized and qualified, such as the MRC-5 or the WI-38 cells, which are accepted as being non-tumorigenic in official compendia (Table 2) [3,65]. Such considerations contrast with the need for iterative testing when working with solid organs or with pooled batches of donor cells, which constitutes a main driver of direct cell-manufacturing costs [20]. Furthermore, when adopting a homologous and allogeneic therapeutic approach using defined primary progenitor-cellular APIs, the implementation of standardized cytotherapeutic protocols is made possible for multi-centric patient treatment, via the deposit of MCB materials in centralized cell repositories [6,19,20]. This aspect provides additional advantages in terms of API consistency and homogeneity for advanced therapy medicinal products (ATMP) or for standardized transplant products (TrSt) in particular, with a simplified and an appropriate documentation in a single cell-type master file and in related manufacturing procedures [18,54,57,59,86–88].
It is worthy to mention that, despite the proven high utility of diploid-cell substrates in public-health applications and the considerable recent interest in the use of such materials as cellular APIs, highly specific ethical and moral considerations or debates have regularly been placing the subject in or out of the limelight [9–16]. Voiced and documented by bioethicists and religious scholars, such proceedings have historically revolved specifically around the original tissue procurement, necessarily serving for the establishment of primary diploid-cell sources. Indeed, the use of discarded pre-natal tissues has been extensively debated, with implications for wide arrays of people involved with the derived products (e.g., physicians administering vaccines produced with diploid-cell substrates) [14,15]. Such fundamental and applied considerations may be interpreted in various ways, yet they do not appear to bare incidence on the relatively elevated rates of pregnancy termination in Western societies and the quantifiable global health gains procured by products of the biotechnological industry [7,16,70,91]. Furthermore, it should be pointed out again that modern frameworks and workflows for the original tissue procurement and for derived biotechnological applications have been regularly updated, to assure conformity with medical ethics and the optimal guaranteeing of patient or of donor rights [2,54,55,57,92]. Therefore, it may be stated overall that modern primary diploid-cell sources are established only after careful consideration of multiple aspects based on the available historical hindsight, ranging from scientific, technical, ethical, regulatory, and quality endpoints [1,2].
5. Formulation Possibilities for Therapeutic Primary Progenitor Cells in Tissue Engineering and in Regenerative Medicine
Various global hurdles currently affect both the transposition and the translation steps during the development of cell-based or of cell-derived therapeutic products, which mainly revolve around the costs of cellular API manufacture and the complexity of regulatory submissions [30–37]. In view of applying banked primary progenitor cells in regenerative medicine, specific technical and mechanistic considerations guide the choice of the development pathways for finished therapeutic products to eventually be clinically applied. Therefore, process-based optimization and quality-oriented approaches to product design and development are current cornerstones for the efficient provision of cellular APIs and products [20,51]. As previously mentioned, specific primary progenitor-cell sources may be considered as optimally compatible with such industrial-scale cGMP cell-manufacturing considerations, due to the favorable inherent technical and biological characteristics (Figure S2) [93,94].
Notably, primary progenitor-cell types recently established under the Swiss progenitor-cell-transplantation program have been thoroughly studied in terms of manufacturing optimization and of formulation possibilities for various clinical applications in regenerative medicine [1,41,43,47,52,53]. In particular, it was technically demonstrated that the final forms of the cellular APIs could be considered as homogenous and stable in view of viable cell-based products administration (i.e., cryogenic storage of the final API lots) or the development of treatments based on devitalized cells and on cell derivatives (i.e., cell lyophilizates to be stored at refrigerated temperatures) (Figure 4) [20,51]. Therefore, major technical and logistical gains may potentially be procured by the use of standardized and stable progenitor-cell-based APIs in diversified therapeutic-product applications [51]. Such considerations may be further extended when reviewing the current state-of-the-art in tissue engineering and in regenerative medicine, where a strong focus is set on the study of sub-cellular fractions or of purified cellular byproducts (e.g., exosomes and cell secretomes) for the promotion of tissue repair or of regeneration processes [95,96].
Specific API dosing and pharmacokinetic considerations reveal additional and multifactorial advantages of adopting local homologous treatment-administration options for allogeneic tissue-specific primary progenitor cells [1,41,94]. Indeed, the high sustainability of cytotherapeutic product manufacture may technically be further potentiated by the use of relatively low progenitor-cell API doses (i.e., 0.5 × 106 to 3 × 106 cells/product unit), as compared to selected stem-cell-infusion protocols (e.g., 108 to 109 cells/treatment) [20,94]. Therefore and as reported, the pragmatic exploitation of multi-tiered progenitor-cell biobanks may potentially enable the derivation of several billion therapeutic product doses from a single cell source [20,66]. Additionally, the use of primary allogeneic cells, which may be reconstituted on-demand and formulated to suit multiple patient and clinician needs, may potentially and tangibly alleviate the latency existing in autologous cell-therapy approaches, entailed by the lengthy patient-specific cell isolation and cell-culture steps [41,97]. Standardized end-products may then be rapidly available in an off-the-freezer or off-the-shelf setting [51,97].
Within the final tissue-engineering-product (TEP) development, critical importance is set on the choice of the appropriate delivery vehicle or the scaffold material to be used in conjunction with the cellular or cell-based API [1,39,42]. Indeed, in clinical cases where volumetric tissue loss has occurred (e.g., sharp force trauma or blunt force trauma inflicted to or sustained by tendon, muscle, or bone tissues), the therapeutic transplant product must comprise a structural replacement function, characterized by tissue-specific mechanical and biological properties [47,48,50,53]. Additionally, appropriate cytocompatibility must be ensured with the therapeutic exogeneous cells, and optimal biocompatibility must be ensured for the final product implantation in the patient [2,98]. Alternatively, in clinical cases where sub-critical internal tissue defects or topical wounds are present, the delivery vehicle or the cell scaffold may be chosen based on specific functionalities and on relatively simple therapeutic-product-administration modalities [53].
Based on drastic technological advances in the fields of polymers and biomaterials, many options are currently available for the development of novel bioengineering solutions, providing high plasticity and tunability to therapeutic-combination-product prototypes [39,42]. Therein, collagen- or hyaluronic-acid-based scaffolds and vehicles are described as simple and optimally adapted for the product formulation of skin- or tendon-tissue-derived primary progenitor cells, respectively, for the provision of topical or of injectable final preparations (Figure 4) [41,53]. Conversely, homologous tissue-engineering therapeutic applications where load-bearing is important favor the use of porous polymeric scaffolds to be surgically implanted (e.g., progenitor bone-cell-laden constructs) after appropriate conditioning of the cellular components (Figure 4) [1,50]. Apart from the choice of the appropriate scaffold or vehicle for a given application, numerous manufacturing parameters must be taken into account during the combination-product development phases, such as the method of cell seeding, the specifications of product pre-implantation incubation, the need for mechanical- or chemical-construct stimuli, etc. [1].
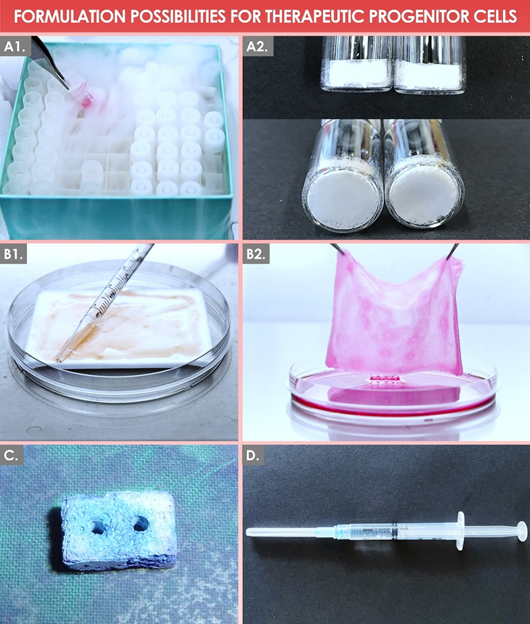
Figure 4. Graphical illustrative overview of selected formulation possibilities for therapeutic primary-progenitor cells in regenerative medicine. (A1) Final form of a cellular API stored as viable cell units in liquid nitrogen. (A2) Final form of a cellular API stored as devitalized cells in lyophilizate units at refrigerated temperatures. (B1, B2) Final product form of a PBB early-wound-coverage solution, composed of viable dermal progenitor fibroblasts (e.g., FE002-SK2 cell type) extemporaneously reconstituted on a collagen sheet scaffold, represented before and after overnight in vitro incubation, respectively. (C) Polymer-based solid tissue construct seeded with cultured primary bone progenitor cells (e.g., FE002-Bone cell type). (D) Hyaluronic-acid-based injectable hydrogel product loaded with cultured primary progenitor tenocytes (e.g., FE002-Ten cell type). API, active pharmaceutical ingredient; PBB, progenitor biological bandage.
Furthermore and importantly, the selection of optimal formulation options for the therapeutic progenitor cells depends on considerations around the mechanism of action, which is principally exerted to obtain the claimed therapeutic effect [71,86]. However, the exact elucidation of pharmacodynamic parameters and precise mechanisms of action of allogeneic-cell therapies currently remains incomplete. It has been established that exogeneous cellular materials (i.e., structural proteins, soluble factors, and enzymes) may act upon the host or recipient through complex and additive or synergistic paracrine modulation of the pathways and the processes of repair and regeneration (Figure S2) [20]. Further research around the mechanisms of action and the related clinical effects of products containing progenitor-cell APIs shall further contribute to optimize and orient the choice of product excipients and may provide information leading to adapted modes and regimens of administration [41,51].
6. Regulatory Considerations for Therapeutic Primary Progenitor Cells and for Related Regenerative Medicine Products
As regards regulatory classification, therapeutic products containing or consisting of cultured progenitor cell APIs are considered as TrSt products in Swiss laws and regulations or as (combined) ATMPs in European texts, notably due to the in vitro cell-expansion steps, which constitute substantial manipulations (Table 5) [58,59,86–88].
Table 5. Definitions pertaining to the various categories of therapeutic products containing or consisting of therapeutic cells in applicable Swiss and European laws and regulations [59,86–88]. ATMP, advanced therapy medicinal product; cATMP, combined advanced therapy medicinal product; CH, Helvetic Confederation; EU, European Union; TEP, tissue-engineering product; TrSt, standardized transplant product.
|
Product Category (Jurisdiction) |
Therapeutic-Product Definition Excerpts 1 |
|
ATMP (EU) |
A medicinal product for human use consisting of (…), a somatic cell therapy medicinal product, or a tissue-engineered product. Somatic-cell-therapy medicinal product means a biological medicinal product that contains or consists of cells or tissues that have been subject to substantial manipulation so that the biological characteristics, physiological functions, or structural properties relevant for the intended clinical use have been altered, or of cells or tissues that are not intended to be used for the same essential function(s) in the recipient and the donor and is presented as having properties for, or is used in or administered to human beings with a view to treating, preventing, or diagnosing a disease through the pharmacological, immunological, or metabolic action of its cells or tissues. |
|
cATMP (EU) |
An ATMP that must incorporate, as an integral part of the product, one or more medical devices (…), and its cellular or tissue part must contain viable cells or tissues, or its cellular or tissue part containing non-viable cells or tissues must be liable to act upon the human body with an action that can be considered as primary to that of the devices (…). |
|
TEP (EU) |
A product that contains or consists of engineered cells or tissues, and is presented as having properties for, or is used in or administered to human beings with a view to regenerating, repairing, or replacing a human tissue. A tissue-engineered product may contain cells or tissues of human or animal origin, or both. The cells or tissues may be viable or non-viable. It may also contain additional substances, such as cellular products, bio-molecules, biomaterials, chemical substances, scaffolds, or matrices. Products containing or consisting exclusively of non-viable human or animal cells and/or tissues, which do not contain any viable cells or tissues and which do not act principally by pharmacological, immunological, or metabolic action, shall be excluded from this definition. |
|
TrSt (CH) |
A transplant product that is intended for transfer to a human and/or whose production process can be standardized, which consists of, or contains, autogenous, allogeneic, or xenogeneic vital organs, tissues, or cells and which is manufactured by means of a standardized procedure. When transplanted, these organs, tissues, or cells are generally manipulated in such a way that their original biological characteristics, physiological functions, or structural properties are affected, or the cells or tissues are not intended to perform essentially the same function(s) in the recipient as in the donor. These can be products from somatic-cell therapy (…) or tissue engineering. Among other aspects, the transplant product serves to regenerate, improve, or influence the human physiological body functions by means of a pharmacological, immunological, or metabolic effect on humans, or it can be used to replace human tissue in order to heal or protect against illnesses, injuries, or impairments. |
1 Definitions have been summarized to present the parameters and the requirements applicable to therapeutic products based on cultured primary progenitor-cell technology, which include by definition a substantial manipulation (i.e., in vitro culture expansions) of the therapeutic cellular materials [1,35,97].
Current applicable laws and regulations, guidelines, or guidance and reference documents and texts for cell-based therapeutic products mainly insist on the risk analysis-based optimization of material sourcing and of subsequent manufacturing processes, in order to guarantee product quality and patient safety [51,54,57,61,63,64,80,85]. Therefore, industrial-product manufacturers individually bear the responsibility of defining, within the provided normative frameworks and workflows, all the technical aspects related to specific cellular-API and cytotherapeutic-product preparation, including processes, specifications and quality attributes, controls, target and acceptance criteria, and specific risk-mitigation-oriented reasoning [51,63,64,80,81]. Such considerations are derived from the classical quality-assurance systems developed for pharmaceutical industries, historically tasked with the manufacture of small-molecule therapeutic-drug products [80,81].
The recent and global introduction of specific cytotherapeutic-product classifications and of related requirements relative to manufacturing have been documented to drastically restrict the number of cell-based or cell-derived products reaching the market (Table 5) [30–34]. Specifically, university hospitals in particular have had to adapt the existing therapeutic practices comprising or consisting of cell therapies (e.g., preparation of autologous keratinocyte sheets for burn patients), in order to comply with the applicable legal frameworks [35,41,42,97]. While the quality-oriented technical and manufacturing aspects of cellular therapies administered to in-house patients have been updated and adapted to be compatible with GMPs, various approaches have been adopted with regard to finished-product or therapy registration or authorization [41,97]. Indeed, the dynamic proposition of innovative cell-based therapies by academic centers and public institutions may often not be compared to the development of similar products by private industries. Therefore, numerous alternative regulatory pathways have been proposed or implemented instead of product-market authorizations, for the maintenance of the clinical use of proven cellular therapies in public hospitals and burn centers in particular [97].
Importantly, based on best practices in the pharmaceutical industry and on specific requirements for the processing of transplant materials, high importance is set on the constitution of cellular-API or cellular-product master files [54–57]. Therefore, summarized compilations of comprehensive data available for a given cellular or cell-derived API and for the corresponding finished products may be prepared in tabular form, similarly to the structure of investigational medicinal-product dossiers (IMPD) [20]. Such recording of the constantly evolving body of knowledge around specific progenitor-cell sources enables the up-to-date and demonstrable insurance of API safety and quality (Table S1) [1,51]. Alternatively, general or specific cellular-API monographs may be constituted by local centers or regional institutions, enabling a conservative transition with regard to quality specifications upon cell-source renewal, manufacturing-scheme updates, or eventual technology transposition (Supplementary Documents S1 and S2) [6,20,57,93]. Furthermore and specifically for cytotherapies which benefit from historical documentation of safety and effectiveness, a cellular-product monograph as defined by the EDQM may be established and adapted according to the updated manufacturing processes or to the gathered clinical evidence (Table S2) [57]. Overall, the constitution of such local documentary records and working tools by public hospitals is in line with quality-assurance guidelines and the related pursuit of risk minimization (Table S3) [51,97]. Most importantly and even if such local or internal documentation is only applicable to individual clinical institutions, careful devising and iterative updates of such data and reference texts by the custodian of records or by the responsible person are essential for the appropriate transmission of knowledge between professional generations, in parallel to the evolution of specific know-how [20,97].
Furthermore, to ensure the widespread applicability or the eventual recognition of processes and of related cellular API monographs established by individual institutions, various specific homologation methods exist, among which is the creation of a tissue or cell monograph (i.e., defined tissue- and cell-based preparations, with specified applications, historically proven as clinically safe and effective) [57]. While this pathway is potentially well-adapted for finished products and ready-to-use tissue- or cell-based preparations, the homologation of cellular API types may be performed with the inclusion of general or specific monographs in recognized compendia, such as a pharmacopoeia. For example, a recent entry in the European Pharmacopoeia (Ph. Eur.) described human hematopoietic stem cells for therapeutic application in the ad hoc monograph “Cellulae stirpes haematopoietica humanae,” attesting to the transition toward the inclusion of cell-based APIs in official lists of pharmaceutical standards [99].
Specifically, the process- and quality-related parameters around diploid-progenitor-cell sources may be easily transposed from the existing practices around biotechnological substrates, as most aspects of cell sourcing, processing, and manufacturing are conserved in the case of cellular-API production [18,57,60–62,65]. However, due to the extensive time required for the generation of data, the review of data, and the approval of specific texts in legally binding documents and compendia, the importance of local and decentralized gathering of the evolving documentation appears as essential for the preservation of knowledge around existing cell therapies. On a pragmatic and practical front, many groups have proposed the return of the magistral preparation of TEPs and ATMPs in public hospitals, mirroring practices in phage therapy [97,100,101]. Therefore, while quality prerequisites and cGMP manufacturing may be implemented for the centralized or local (i.e., point-of-care manufacturing) production of cell-based therapies, sufficient medical-prescription flexibility would be provided to clinicians, while ensuring local adequation with legal requirements [20,93,102]. Overall, fine integration and interpretation of the applicable laws and regulations should be included in all phases of design and development of novel cell-based or cell-derived therapeutic products, for the optimization and rationalization of both risks and costs [20,51].
7. Preclinical and Clinical Experience with Therapeutic Primary Progenitor Cells in the Swiss Progenitor-Cell-Transplantation Program
Originally implemented in the 1990s, the Swiss progenitor-cell-transplantation program has served as a functional translational basis for the continued allogeneic therapeutic applications (i.e., preclinical and clinical) of human primary progenitor cells [1,2]. It was specifically outlined and reported that such tissue-specific cell types, adequately and parallelly processed for in vitro culture initiation from regulated organ donations, presented considerable potential for applications in homologous regenerative medicine (Table 6, Figures 5 and S8) [2,20,39].
Table 6. Overview of the various tissue-specific homologous applications considered or investigated at preclinical and clinical stages under the Swiss progenitor-cell-transplantation program [1]. It may be noted that initial transposition of FE002-derived progenitor-cell sources to GMP manufacture were performed in collaboration and under contract with BioReliance in the UK (Merck group, Glasgow and Stirling, UK). Therefore, in view of GMP cell banking for API manufacture, most of the control and testing schemes were based on requirements existing for vaccine substrates, such as presented in Table 2. ECACC, European collection of authenticated cell cultures; FIRDI, Food industry research and development institute; GMP, good manufacturing practices; NA, non-applicable; UK, United Kingdom.
|
Starting Tissue Types |
Progenitor-Cell-Type Examples |
Research, Preclinical, and Clinical Application Work and Milestones |
Cell-Type Deposit References |
|
Skin |
FE002-SK2 1 |
Manufacturing: Industrial GMP cell-manufacturing upscaling and international transposition [20,51]. Clinical trials: Severe burns, refractory cutaneous ulcers, and skin donor-site wounds [38,40,41,43]. |
ECACC 12070301-FE002-SK2; FIRDI BCRC 960460 |
|
Cartilage |
FE002-Cart 1 |
Manufacturing: Industrial cell-banking and cell-manufacturing optimization [52]. Preclinical studies: Safety of cell transplantation in caprine model [52]. |
ECACC 12070303-FE002-Cart; FIRDI BCRC 960459 |
|
Tendon |
FE002-Ten 1 |
Manufacturing: Industrial cell-banking and cell-manufacturing optimization [53,94]. Preclinical studies: Safety of cell transplantation in lagomorph model [53]. |
ECACC 12070302-FE002-Ten; FIRDI BCRC 960461 |
|
Bone |
FE002-Bone |
Manufacturing: Optimized cell banking and cell manufacturing [50]. Preclinical studies: Safety of cell transplantation in rat models [103]. |
NA |
|
Muscle |
FE002-Mu |
Manufacturing: Optimized cell banking and cell manufacturing [48]. Preclinical studies: Safety of cell transplantation in murine model [104]. |
NA |
|
Intervertebral disc |
FE002-Disc |
Manufacturing: Optimized cell banking and cell manufacturing. |
NA |
|
Lung |
FE002-Lu |
Manufacturing: Optimized cell banking and cell manufacturing. |
NA |
1 The mechanically isolated FE002-SK2, FE002-Cart, and FE002-Ten primary progenitor-cell types were deposited with the ECACC (UK) and the FIRDI (Taiwan) centralized-cell repositories in 2012 [105].
Specifically, the reported clinical applications of banked human progenitor cells in regenerative medicine have been organized under a dedicated cell-transplantation program since 1991, which was registered with the Swiss Federal office of public health and with the Swiss therapeutic products agency (i.e., Swissmedic) [1,2,28,35]. From a translational point of view, the most research and clinical experience has been generated over the past three decades in the Lausanne University Hospital around the therapeutic applications of skin-derived primary progenitor fibroblasts [20,38–43]. The simplicity of manufacturing and the high consistency of such cellular APIs (e.g., FE002-SK2 progenitor-cell type) have enabled the efficient and continued preparation of clinical-grade cell batches by the in-house GMP platform (Table S1) [20,97]. Thereafter, various pharmaceutical forms of the topical finished products have been studied and were therapeutically used for managing pediatric burn wounds, skin donor-site wounds, and geriatric cutaneous ulcers in particular, yielding highly encouraging results (Table S2, Figure S8) [38–43].
In detail, the on-demand extemporaneous and direct off-the-freezer seeding of viable dermal progenitor fibroblasts on collagen-sheet scaffolds have enabled the rapid and simple preparation of the progenitor-biological-bandage (PBB) early-wound coverage solution (Tables S2 and S3; Figures 5 and S8) [38,41]. Initially described in the Lancet in 2005 for applications in pediatric burn victims, clinical applications of PBBs have been continuously performed up to the present day and have recently been authorized by Swissmedic (i.e., for clinical trial use) in the preparative context of a new prospective clinical trial (i.e., authorization under Swissmedic reference N° 2020TpP1010) [38,97]. The PBB products are iteratively applied by clinicians during the burn-patient bandage-exchange procedures and therefore do not require stappling. In the context of three distinct clinical trials, the therapeutic use of PBBs has been reported to enable an optimal and rapid skin-reconstruction promotion, allowing for restoration of high elastic properties and skin-pigmentation balance [38,41,43]. In addition, notable reductions in local pain, scar hypertrophy, tissue retraction and inflammation, or the need for additional skin autografting were documented [41].
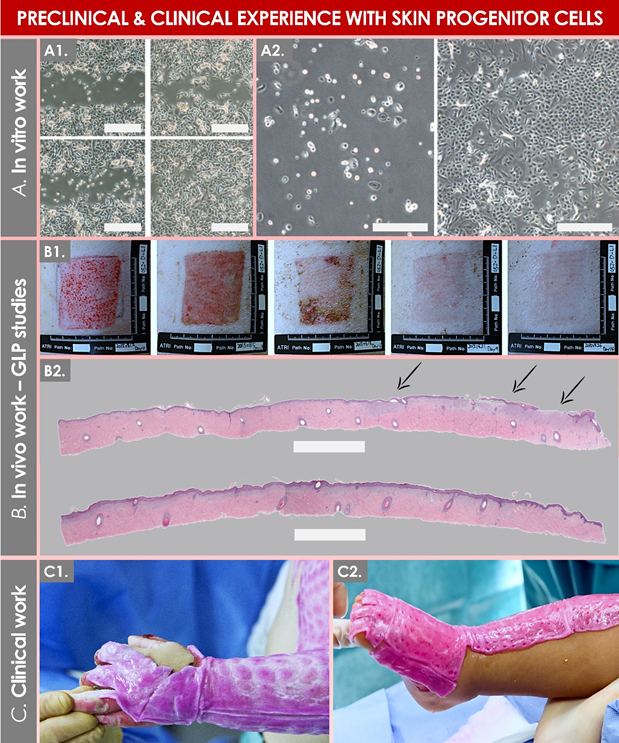
Figure 5. Illustrative overview of preclinical and clinical experience gained with primary progenitor dermal fibroblasts (e.g., FE002-SK2 cell source). (A1) Results of a keratinocyte migration assay showing an increased gap closure at the 9 h timepoint (bottom quadrants) versus the 6 h timepoint (upper quadrants), with an increased relative gap-closure rate in the treatment (i.e., progenitor cell-conditioned medium) group (right quadrants). Scale bars = 50 µm. (A2) Results of a co-culture assay showing the positive effect of 6 × 103 progenitor fibroblasts (right panel) on the proliferation of primary keratinocytes after 6 days of co-culture, versus keratinocytes cultured alone (left panel). Scale bars = 50 µm. (B1) Photographic evolutive results of a wound-healing assay in a GLP porcine skin-excision model treated with a hydrogel containing progenitor fibroblasts over 14 days. (B2) Histology results of the GLP porcine wound-healing assay, showing epidermal detachment in the sham group (upper portion, evidenced by arrows) and relatively enhanced tissue repair in the cellular-therapy group (lower portion). Scale bars = 750 µm. (C1, C2) Photographic imaging of pediatric burn patients presenting limb cutaneous wounds and treated with progenitor biological bandages. GLP, good laboratory practices. Modified and adapted with permission from Laurent et al., 2020, 2021 [20,41].
Technically, the validity of the adopted methodology for the primary progenitor-cell sourcing and for subsequent cell banking as described under the Swiss progenitor-cell-transplantation program was confirmed, notably following the approval of use of the FE002-SK2 cell source in specific clinical trials (i.e., authorized to date by the FDA, the TFDA, and the PMDA) [20]. Therefore and importantly, over twenty years of clinical applications in various clinical studies have enabled to conclude to both the safety and the positive therapeutic effects procured by PBB constructs, notably in phase I and II clinical trials in Switzerland, while additional trials are planned locally and are also performed around the same cell source throughout Asia (e.g., ClinicalTrials.gov identifiers: NCT02737748, NCT03624023) [20]. Based on the obtained encouraging clinical results in cutaneous regenerative medicine, subsequent efforts and resources have been allocated toward tissue-specific therapeutic applications of primary progenitor cells in musculoskeletal and in soft-tissue regenerative medicine (Table 6) [1,93]. Notably, recent progress in bioengineering approaches for the treatment of cartilage, bone, and tendon lesions have provided important technical bases for the further preclinical and clinical study of diverse and robust tissue-specific primary progenitor-cell sources [50,52,53]. Therefore, the endgame of the Swiss progenitor-cell-transplantation program consists in providing a holistic, solid, durable, and transposable cytotherapeutic rationale of a tissue-specific allogeneic primary progenitor-cell-based approach in highly specialized regenerative medicine [1].
8. Conlusion
Based on the unique biological and technical specificities of primary diploid progenitor cells and on the available historical experience around the safe industrial and clinical uses thereof, it is of extremely high current interest to further investigate specific cell sources for the development of novel biotechnological substrates and allogeneic-cell-therapy products. The existing high demand for appropriate biotechnological substrates is currently illustrated by the global COVID-19-pandemic context. Furthermore, the elevated and increasing numbers of patients presenting acute or degenerative affections prompt the study of novel therapeutic-product types, such as cell-based or cell-derived solutions for regenerative medicine. Therefore, it may be overall assessed and stated that selected modern primary progenitor-cell types, adequately sourced and established, will potentially be applied with high versatility as industrial biotechnological platforms and/or as high-quality APIs in cellular therapies and in tissue-engineering products. Such diversified approaches may potentially further contribute to modern therapeutic-product development efforts and tangibly support the implementation of effective novel solutions for patient therapeutic management.
Abbreviations
|
API |
Active pharmaceutical ingredient |
|
ATCC |
American-type culture collection |
|
ATMP |
Advanced-therapy medicinal product |
|
cATMP |
Combined advanced-therapy medicinal product |
|
CD |
Cluster of differentiation |
|
cGMP |
Current good manufacturing practices |
|
CH |
Helvetic Confederation |
|
CHUV |
Centre hospitalier universitaire vaudois |
|
CMV |
Cytomegalovirus |
|
DMEM |
Dulbecco’s modified Eagle medium |
|
DMSO |
Dimethyl sulfoxide |
|
DNA |
Deoxyribonucleic acid |
|
EBV |
Epstein–Barr virus |
|
EC |
European Commission |
|
ECACC |
European collection of authenticated cell cultures |
|
ECB |
Extended cell bank |
|
ECM |
Extra-cellular matrix |
|
EDQM |
European Directorate for the Quality of Medicines & Healthcare |
|
EDTA |
Ethylenediaminetetraacetic acid |
|
EMA |
European Medicines Agency |
|
EOPCB |
End-of-production cell bank |
|
EU |
European Union |
|
FBS |
Fetal bovine serum |
|
FDA |
US Food and Drug Administration |
|
FIRDI |
Food Industry Research and Development Institute |
|
FOPH |
Swiss Federal Office of Public Health |
|
FPC |
Fibroblast progenitor cell |
|
GLP |
Good laboratory practices |
|
GMP |
Good manufacturing practices |
|
HACCP |
Hazard analysis critical control point |
|
HAV |
Hepatitis A virus |
|
HBoV |
Human bocavirus |
|
HBV |
Hepatitis B virus |
|
hCMV |
Human cytomegalovirus |
|
HCV |
Hepatitis C virus |
|
HHV |
Human herpes virus |
|
HIV |
Human immunodeficiency virus |
|
HLA |
Human leucocyte antigen |
|
HPV |
Human papillomavirus |
|
HTLV |
Human T-cell lymphotropic virus |
|
HuPyV |
Human polyomavirus |
|
ICH |
International Council for Harmonization |
|
IMPD |
Investigational medicinal product dossier |
|
IPC |
In-process control |
|
ISO |
International Organization for Standardization |
|
KIPyV |
KI polyomavirus |
|
MCB |
Master cell bank |
|
MD |
Medical device |
|
MoA |
Mechanism of action |
|
MRC |
Medical research council |
|
NAT |
Nucleic-acid amplification techniques |
|
PBB |
Progenitor biological bandage |
|
PCB |
Parental cell bank |
|
PD |
Population doublings |
|
PDT |
Population-doubling time |
|
PDV |
Population-doubling value |
|
Ph. Eur. |
European pharmacopoeia |
|
PPC |
Post-process control |
|
PRC |
People’s Republic of China |
|
QA |
Quality assurance |
|
QC |
Quality control |
|
QFPERT |
Quantification of reverse-transcriptase activity by ultracentrifugation and quantitative fluorescent product-enhanced reverse transcriptase |
|
RH |
Relative humidity |
|
RT-PCR |
Real-time polymerase chain reaction |
|
SFPC |
Skin-fibroblast-progenitor cells |
|
SV40 |
Simian virus 40 |
|
TEM |
Transmission electron microscopy |
|
TEP |
Tissue-engineering product |
|
TrSt |
Standardized transplant product |
|
UK |
United Kingdom |
|
USA |
United States of America |
|
WCB |
Working cell bank |
|
WHO |
World Health Organization |
|
WI |
Wistar Institute |
|
WUPyV |
Wu polyomavirus |