Chronic inflammatory diseases and transplant rejection represent major challenges for modern health care. Thus, identification of immune checkpoints that contribute to resolution of inflammation is key to developing novel therapeutic agents for those conditions. In recent years, the CD83 (cluster of differentiation 83) protein has emerged as an interesting potential candidate for such a “pro-resolution” therapy. This molecule occurs in a membrane-bound and a soluble isoform (mCD83 and sCD83, respectively), both of which are involved in resolution of inflammation. Originally described as a maturation marker on dendritic cells (DCs), mCD83 is also expressed by activated B and T cells as well as regulatory T cells (Tregs) and controls turnover of MHC II molecules in the thymus, and thereby positive selection of CD4+ T cells. Additionally, it serves to confine overshooting (auto-)immune responses. Consequently, animals with a conditional deletion of CD83 in DCs or regulatory T cells suffer from impaired resolution of inflammation. Pro-resolving effects of sCD83 became evident in pre-clinical autoimmune and transplantation models, where application of sCD83 reduced disease symptoms and enhanced allograft survival, respectively.
- CD83
- resolution of inflammation
- IDO/TGF-β-axis
- pro-resolving macrophages
- transplantation
1. Introduction
2. CD83: From Maturation Marker to Pro-Resolving Checkpoint Molecule
Since its discovery in 1992 as a surface molecule on activated immune cells such as DCs and B cells, the CD83 molecule has been intensively studied and characterized [18][19][20]: the CD83 protein is highly conserved among distinct species as murine, and human CD83 share 63% amino acid identity [21][22]. Membrane-bound CD83 (mCD83) is extensively glycosylated, which almost doubles its theoretical molecular weight from 23 kDa to 45 kDa, and it consists of three domains: an extracellular Ig-like domain, a transmembrane domain and a cytoplasmic domain [18]. A soluble isoform (sCD83) that consists largely of the extracellular domain is released into the supernatant of activated DCs and B cells [23]. This isoform is generated either by alternative splicing or by proteolytic cleavage of mCD83 [23][24]. The sCD83 molecule is present in low levels in sera of healthy donors, but its abundancy increases in sera of patients with hematological malignancies, e.g., multiple myeloma and acute myeloid leukemia, where it inversely correlates with progression-free survival [25]. In addition, patients with chronic inflammatory diseases, such as multiple sclerosis [26] and rheumatoid arthritis [27] show elevated sCD83 serum levels. Originally described as a marker for mature DCs (mDCs) [22][28][29][30][31], CD83 expression has been demonstrated on many other activated immune cells, including neutrophils [32], monocytes, macrophages (Mφ) [33], B/T cells [34], NK cells [35], and Tregs [36]. Moreover, also non-immune cells like epithelial cells of the thymus, airways, and intestine express CD83 [37][38][39]. Interestingly, CD83 is also a target of immune escape mechanisms. For instance, infection of DCs with herpes viruses results in proteasomal degradation of CD83, which leads to inhibition of potent antiviral immune responses [40][41][42]. Furthermore, Hodgkin lymphoma cells express CD83 to subvert anti-tumor T cell responses, in part by secretion of sCD83 [43]. In the past few years, various studies have elucidated the biological role of the mCD83 molecule in the context of homeostasis and immune pathologies. Within these studies, the mCD83 protein has emerged as a master regulator for CD4+ T cell development [44], and recently mCD83 was characterized as an immunoregulatory molecule, which contributes to maintenance of tolerance [30][36]. Moreover, the sCD83 isoform possesses striking capacities to induce the resolution of inflammation, shown in different pre-clinical models for chronic inflammatory/autoimmune diseases, food allergy and transplantation [39][45][46][47][48][49][50][51]. Within the following sections, we will summarize current knowledge on CD83 elicited signaling events and its pro-resolving function in homeostasis, autoimmune pathologies as well as in transplantation.2.1. Biological Function of CD83 and Its Induced Signaling Events
2.2. Role of mCD83 in the Resolution of Inflammation
As mentioned above, the deleterious effect of complete CD83-deletion on CD4+ cell development has impeded clear predictions of the biological function of CD83 in inflammation for a long time. Due to their lack of peripheral CD4+ T cells, CD83−/− mice show reduced responses in a contact hypersensitivity model, which is dependent on proper T cell reaction [44][52], and their remaining T cells are hyperresponsive to stimulation [52]. Thus, employing these mice does not allow evaluation of the relevance of CD83 expression on different cell types for immune responses. To circumvent this problem, we generated mice where CD83 can be deleted by the Cre-LoxP system to enable investigations on its cell-specific biologic functions. Preliminary studies have revealed that CD83 deletion in B cells interferes with the proper formation of germinal center reaction and antibody production in response to bacterial infection [53]. Further data on conditional deletion of CD83 in DCs and Tregs have disclosed its vital role for the resolution of inflammation, which we will discuss in the following section.3. Clinical Relevance of sCD83 for Therapeutic Purposes
There is an extensive body of evidence, demonstrating that sCD83 administration is a potent means to promote pro-resolution effects in preclinical disease models. Thus, in the following chapter, we will summarize the beneficial effects induced by sCD83 in these models, with a special focus on chronic inflammatory and autoimmune conditions as well as strategies to prevent organ transplant rejections.3.1. sCD83 Promotes the Resolution of Chronic Inflammation
3.2. sCD83 Prevents Graft Rejection by Induction of Tolerogenic Mechanisms
In contrast to autoimmune diseases, in which the immune system reacts to self-antigens, unwanted inflammatory responses of innate and adaptive immune cells occur in the field of transplantation. Rejections of allografts occur due to fatal immune reactions of the recipient to the donor tissue. These inflammatory responses rely on differences of highly polymorphic MHC molecules between recipient and donor, resulting in tissue damage and finally rejection of the transplanted tissue. Current therapeutic approaches in patients receiving organ transplants often rely on a non-specific immunosuppressive medication, such as glucocorticoids, cytostatics, calcineurin inhibitors, or mTOR inhibitors with all known associated negative side effects [54]. Thus, patients after organ transplantation often suffer from drug-associated toxicity, reduced resistance to infections and development of malignancies. Consequently, new therapeutic agents, which establish or induce immune tolerance, promote tissue repair, contribute to resolution of inflammation are urgently needed. For this, researchers pursue amongst others the following strategies: (i) induction or transfer of Tregs, which are able to induce immune tolerance, and (ii) modulation of APC populations including DCs as well as Mφ towards regulatory cells, which can promote Treg differentiation and thus, induce immune tolerance. In this respect, regulatory Mφ, DCs, as well as Tregs, which are able to resolve inflammatory responses have been used in clinical trials as a cellular therapy in combination with immunosuppressive drugs in kidney transplantation [55].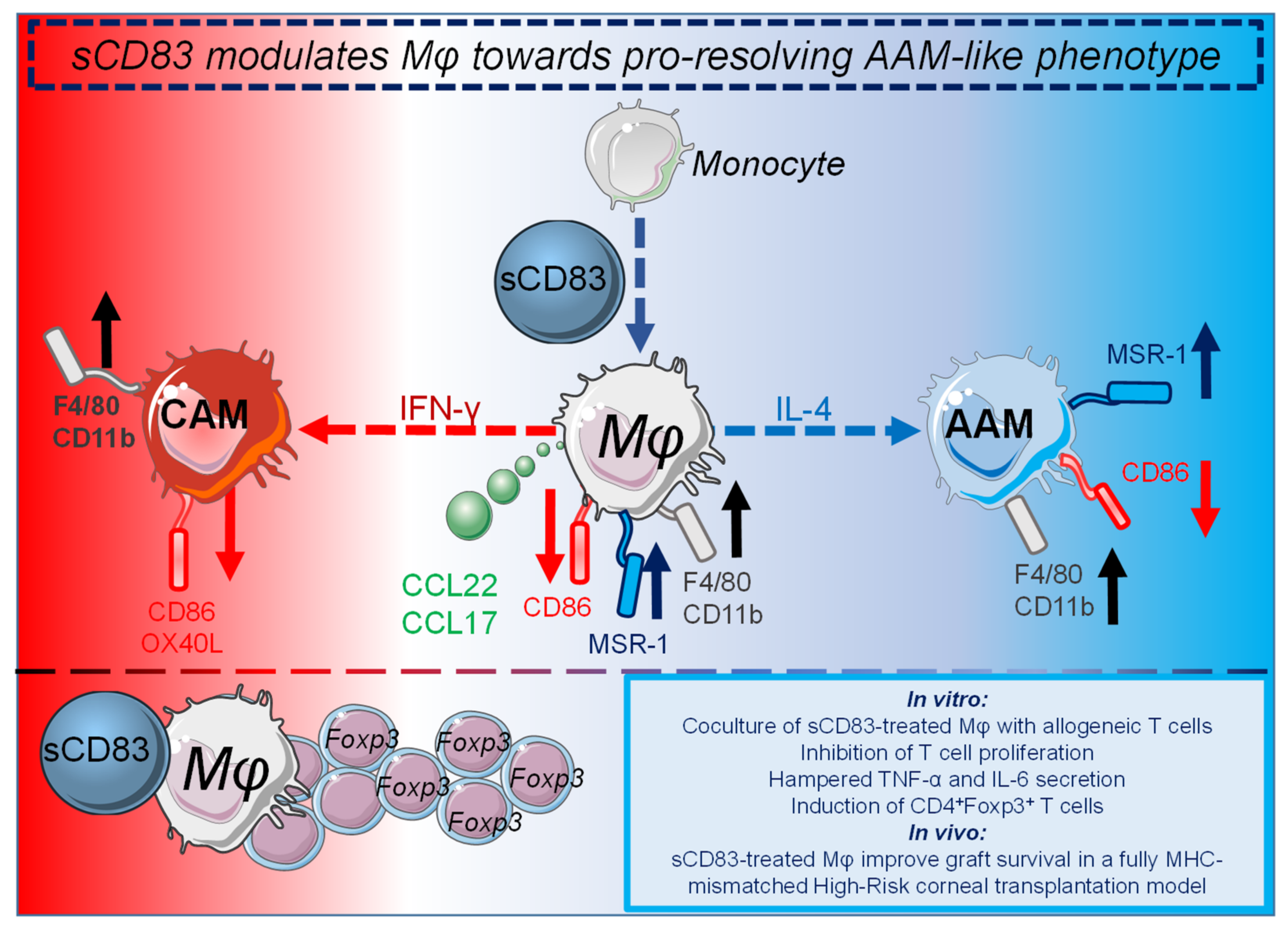 The sCD83 induced pro-resolving changes on Mφ are summarized in Figure 1.
The sCD83 induced pro-resolving changes on Mφ are summarized in Figure 1.
4. sCD83: Conclusions, New Insights and Future Direction
References
- Ortega-Gomez, A.; Perretti, M.; Soehnlein, O. Resolution of inflammation: An integrated view. EMBO Mol. Med. 2013, 5, 661–674.
- Uderhardt, S.; Martins, A.J.; Tsang, J.S.; Lammermann, T.; Germain, R.N. Resident Macrophages Cloak Tissue Microlesions to Prevent Neutrophil-Driven Inflammatory Damage. Cell 2019, 177, 541–555.
- Jones, H.R.; Robb, C.T.; Perretti, M.; Rossi, A.G. The role of neutrophils in inflammation resolution. Semin. Immunol. 2016, 28, 137–145.
- Cifuentes-Rius, A.; Desai, A.; Yuen, D.; Johnston, A.P.R.; Voelcker, N.H. Inducing immune tolerance with dendritic cell-targeting nanomedicines. Nat. Nanotechnol. 2021, 16, 37–46.
- Netea, M.G.; Balkwill, F.; Chonchol, M.; Cominelli, F.; Donath, M.Y.; Giamarellos-Bourboulis, E.J.; Golenbock, D.; Gresnigt, M.S.; Heneka, M.T.; Hoffman, H.M.; et al. A guiding map for inflammation. Nat. Immunol. 2017, 18, 826–831.
- Schett, G.; Neurath, M.F. Resolution of chronic inflammatory disease: Universal and tissue-specific concepts. Nat. Commun. 2018, 9, 3261.
- Watanabe, S.; Alexander, M.; Misharin, A.V.; Budinger, G.R.S. The role of macrophages in the resolution of inflammation. J. Clin. Investig. 2019, 129, 2619–2628.
- Filep, J.G.; Ariel, A. Neutrophil heterogeneity and fate in inflaMed. tissues: Implications for the resolution of inflammation. Am. J. Physiol Cell Physiol. 2020, 319, C510–C532.
- Marwick, J.A.; Mills, R.; Kay, O.; Michail, K.; Stephen, J.; Rossi, A.G.; Dransfield, I.; Hirani, N. Neutrophils induce macrophage anti-inflammatory reprogramming by suppressing NF-kappaB activation. Cell Death Dis. 2018, 9, 665.
- Munoz-Rojas, A.R.; Kelsey, I.; Pappalardo, J.L.; Chen, M.; Miller-Jensen, K. Co-stimulation with opposing macrophage polarization cues leads to orthogonal secretion programs in individual cells. Nat. Commun. 2021, 12, 301.
- Oishi, Y.; Manabe, I. Macrophages in inflammation, repair and regeneration. Int. Immunol. 2018, 30, 511–528.
- Sugimoto, M.A.; Sousa, L.P.; Pinho, V.; Perretti, M.; Teixeira, M.M. Resolution of Inflammation: What Controls Its Onset? Front. Immunol. 2016, 7, 160.
- Sugimoto, M.A.; Vago, J.P.; Perretti, M.; Teixeira, M.M. Mediators of the Resolution of the Inflammatory Response. Trends Immunol. 2019, 40, 212–227.
- Ruiz, F.; Vigne, S.; Pot, C. Resolution of inflammation during multiple sclerosis. Semin. Immunopathol. 2019, 41, 711–726.
- Schett, G. Resolution of inflammation in arthritis. Semin. Immunopathol. 2019, 41, 675–679.
- Mori, D.N.; Kreisel, D.; Fullerton, J.N.; Gilroy, D.W.; Goldstein, D.R. Inflammatory triggers of acute rejection of organ allografts. Immunol. Rev. 2014, 258, 132–144.
- Fullerton, J.N.; Gilroy, D.W. Resolution of inflammation: A new therapeutic frontier. Nat. Rev. Drug Discov. 2016, 15, 551–567.
- Zhou, L.J.; Schwarting, R.; Smith, H.M.; Tedder, T.F. A Novel Cell-Surface Molecule Expressed by Human Interdigitating Reticulum Cells, Langerhans Cells, and Activated Lymphocytes Is a New Member of the Ig Superfamily. J. Immunol. 1992, 149, 735–742.
- Grosche, L.; Knippertz, I.; Konig, C.; Royzman, D.; Wild, A.B.; Zinser, E.; Sticht, H.; Muller, Y.A.; Steinkasserer, A.; Lechmann, M. The CD83 Molecule—An Important Immune Checkpoint. Front. Immunol. 2020, 11, 721.
- Li, Z.; Ju, X.; Silveira, P.A.; Abadir, E.; Hsu, W.H.; Hart, D.N.J.; Clark, G.J. CD83: Activation Marker for Antigen Presenting Cells and Its Therapeutic Potential. Front. Immunol. 2019, 10, 1312.
- UniProt, C. UniProt: A hub for protein information. Nucleic Acids Res. 2015, 43, D204–D212.
- Berchtold, S.; Muhl-Zurbes, P.; Heufler, C.; Winklehner, P.; Schuler, G.; Steinkasserer, A. Cloning, recombinant expression and biochemical characterization of the murine CD83 molecule which is specifically upregulated during dendritic cell maturation. FEBS Lett. 1999, 461, 211–216.
- Hock, B.D.; Kato, M.; McKenzie, J.L.; Hart, D.N. A soluble form of CD83 is released from activated dendritic cells and B lymphocytes, and is detectable in normal human sera. Int. Immunol. 2001, 13, 959–967.
- Dudziak, D.; Nimmerjahn, F.; Bornkamm, G.W.; Laux, G. Alternative splicing generates putative soluble CD83 proteins that inhibit T cell proliferation. J. Immunol. 2005, 174, 6672–6676.
- Hock, B.D.; Haring, L.F.; Steinkasserer, A.; Taylor, K.G.; Patton, W.N.; McKenzie, J.L. The soluble form of CD83 is present at elevated levels in a number of hematological malignancies. Leukemia Res. 2004, 28, 237–241.
- Karampoor, S.; Zahednasab, H.; Etemadifar, M.; Keyvani, H. The levels of soluble forms of CD21 and CD83 in multiple sclerosis. J. NeuroImmunol. 2018, 320, 11–14.
- Hock, B.D.; O’Donnell, J.L.; Taylor, K.; Steinkasserer, A.; McKenzie, J.L.; Rothwell, A.G.; Summers, K.L. Levels of the soluble forms of CD80, CD86, and CD83 are elevated in the synovial fluid of rheumatoid arthritis patients. Tissue Antigens 2006, 67, 57–60.
- Lechmann, M.; Berchtold, S.; Hauber, J.; Steinkasserer, A. CD83 on dendritic cells: More than just a marker for maturation. Trends Immunol. 2002, 23, 273–275.
- Bates, J.M.; Flanagan, K.; Mo, L.; Ota, N.; Ding, J.; Ho, S.; Liu, S.; Roose-Girma, M.; Warming, S.; Diehl, L. Dendritic cell CD83 homotypic interactions regulate inflammation and promote mucosal homeostasis. Mucosal. Immunol. 2015, 8, 414–428.
- Wild, A.B.; Krzyzak, L.; Peckert, K.; Stich, L.; Kuhnt, C.; Butterhof, A.; Seitz, C.; Mattner, J.; Gruner, N.; Gansbauer, M.; et al. CD83 orchestrates immunity toward self and non-self in dendritic cells. JCI Insight 2019, 4.
- Tze, L.E.; Horikawa, K.; Domaschenz, H.; Howard, D.R.; Roots, C.M.; Rigby, R.J.; Way, D.A.; Ohmura-Hoshino, M.; Ishido, S.; Andoniou, C.E.; et al. CD83 increases MHC II and CD86 on dendritic cells by opposing IL-10-driven MARCH1-mediated ubiquitination and degradation. J. Exp. Med. 2011, 208, 149–165.
- Yamashiro, S.; Wang, J.M.; Yang, D.; Gong, W.H.; Kamohara, H.; Yoshimura, T. Expression of CCR6 and CD83 by cytokine-activated human neutrophils. Blood 2000, 96, 3958–3963.
- Cao, W.; Lee, S.H.; Lu, J. CD83 is preforMed. inside monocytes, macrophages and dendritic cells, but it is only stably expressed on activated dendritic cells. Biochem. J. 2005, 385, 85–93.
- Cramer, S.O.; Trumpfheller, C.; Mehlhoop, U.; More, S.; Fleischer, B.; von Bonin, A. Activation-induced expression of murine CD83 on T cells and identification of a specific CD83 ligand on murine B cells. Int. Immunol. 2000, 12, 1347–1351.
- Mailliard, R.B.; Alber, S.M.; Shen, H.; Watkins, S.C.; Kirkwood, J.M.; Herberman, R.B.; Kalinski, P. IL-18-induced CD83+CCR7+ NK helper cells. J. Exp. Med. 2005, 202, 941–953.
- Doebbeler, M.; Koenig, C.; Krzyzak, L.; Seitz, C.; Wild, A.; Ulas, T.; Bassler, K.; Kopelyanskiy, D.; Butterhof, A.; Kuhnt, C.; et al. CD83 expression is essential for Treg cell differentiation and stability. JCI Insight 2018, 3, e99712.
- Von Rohrscheidt, J.; Petrozziello, E.; Nedjic, J.; Federle, C.; Krzyzak, L.; Ploegh, H.L.; Ishido, S.; Steinkasserer, A.; Klein, L. Thymic CD4 T cell selection requires attenuation of March8-mediated MHCII turnover in cortical epithelial cells through CD83. J. Exp. Med. 2016, 213, 1685–1694.
- Yu, Y.; Jin, Q.R.; Mi, Y.; Liu, J.Q.; Liu, Z.Q.; Wang, S.; Liu, Z.G.; Yang, P.C.; Zheng, P.Y. Intestinal Epithelial Cell-Derived CD83 Contributes to Regulatory T-Cell Generation and Inhibition of Food Allergy. J. Innate Immun. 2021, 13, 295–305.
- Mo, L.H.; Luo, X.Q.; Yang, G.; Liu, J.Q.; Yang, L.T.; Liu, Z.Q.; Wang, S.; Liu, D.B.; Liu, Z.G.; Yang, P.C. Epithelial cell-derived CD83 restores immune tolerance in the airway mucosa by inducing regulatory T-cell differentiation. Immunology 2021, 163, 310–322.
- Heilingloh, C.S.; Muhl-Zurbes, P.; Steinkasserer, A.; Kummer, M. Herpes simplex virus type 1 ICP0 induces CD83 degradation in mature dendritic cells independent of its E3 ubiquitin ligase function. J. Gen. Virol. 2014, 95, 1366–1375.
- Heilingloh, C.S.; Grosche, L.; Kummer, M.; Muhl-Zurbes, P.; Kamm, L.; Scherer, M.; Latzko, M.; Stamminger, T.; Steinkasserer, A. The Major Immediate-Early Protein IE2 of Human Cytomegalovirus Is Sufficient to Induce Proteasomal Degradation of CD83 on Mature Dendritic Cells. Front. Microbiol. 2017, 8, 119.
- Grosche, L.; Muhl-Zurbes, P.; Ciblis, B.; Krawczyk, A.; Kuhnt, C.; Kamm, L.; Steinkasserer, A.; Heilingloh, C.S. Herpes Simplex Virus Type-2 Paralyzes the Function of Monocyte-Derived Dendritic Cells. Viruses 2020, 12, 112.
- Li, Z.; Ju, X.; Lee, K.; Clarke, C.; Hsu, J.L.; Abadir, E.; Bryant, C.E.; Pears, S.; Sunderland, N.; Heffernan, S.; et al. CD83 is a new potential biomarker and therapeutic target for Hodgkin lymphoma. Haematologica 2018, 103, 655–665.
- Fujimoto, Y.; Tu, L.; Miller, A.S.; Bock, C.; Fujimoto, M.; Doyle, C.; Steeber, D.A.; Tedder, T.F. CD83 expression influences CD4+ T cell development in the thymus. Cell 2002, 108, 755–767.
- Zinser, E.; Lechmann, M.; Golka, A.; Lutz, M.B.; Steinkasserer, A. Prevention and treatment of experimental autoimmune encephalomyelitis by soluble CD83. J. Exp. Med. 2004, 200, 345–351.
- Starke, C.; Steinkasserer, A.; Voll, R.E.; Zinser, E. Soluble human CD83 ameliorates lupus in NZB/W F1 mice. Immunobiology 2013, 218, 1411–1415.
- Eckhardt, J.; Kreiser, S.; Dobbeler, M.; Nicolette, C.; DeBenedette, M.A.; Tcherepanova, I.Y.; Ostalecki, C.; Pommer, A.J.; Becker, C.; Gunther, C.; et al. Soluble CD83 ameliorates experimental colitis in mice. Mucosal. Immunol. 2014, 7, 1006–1018.
- Lin, W.; Man, X.; Li, P.; Song, N.; Yue, Y.; Li, B.; Li, Y.; Sun, Y.; Fu, Q. NK cells are negatively regulated by sCD83 in experimental autoimmune uveitis. Sci. Rep. 2017, 7, 12895.
- Lin, W.; Buscher, K.; Wang, B.; Fan, Z.; Song, N.; Li, P.; Yue, Y.; Li, B.; Li, C.; Bi, H. Soluble CD83 Alleviates Experimental Autoimmune Uveitis by Inhibiting Filamentous Actin-Dependent Calcium Release in Dendritic Cells. Front. Immunol. 2018, 9, 1567.
- McHugh, J. Soluble CD83: A proresolving mediator in inflammatory arthritis? Nat. Rev. Rheumatol. 2019, 15, 319.
- Royzman, D.; Andreev, D.; Stich, L.; Rauh, M.; Bauerle, T.; Ellmann, S.; Boon, L.; Kindermann, M.; Peckert, K.; Bozec, A.; et al. Soluble CD83 Triggers Resolution of Arthritis and Sustained Inflammation Control in IDO Dependent Manner. Front. Immunol. 2019, 10, 633.
- Garcia-Martinez, L.F.; Appleby, M.W.; Staehling-Hampton, K.; Andrews, D.M.; Chen, Y.; McEuen, M.; Tang, P.; Rhinehart, R.L.; Proll, S.; Paeper, B.; et al. A novel mutation in CD83 results in the development of a unique population of CD4+ T cells. J. Immunol. 2004, 173, 2995–3001.
- Krzyzak, L.; Seitz, C.; Urbat, A.; Hutzler, S.; Ostalecki, C.; Glasner, J.; Hiergeist, A.; Gessner, A.; Winkler, T.H.; Steinkasserer, A.; et al. CD83 Modulates B Cell Activation and Germinal Center Responses. J. Immunol. 2016, 196, 3581–3594.
- Diehl, R.; Ferrara, F.; Muller, C.; Dreyer, A.Y.; McLeod, D.D.; Fricke, S.; Boltze, J. Immunosuppression for in vivo research: State-of-the-art protocols and experimental approaches. Cell Mol. Immunol. 2017, 14, 146–179.
- Sawitzki, B.; Harden, P.N.; Reinke, P.; Moreau, A.; Hutchinson, J.A.; Game, D.S.; Tang, Q.; Guinan, E.C.; Battaglia, M.; Burlingham, W.J.; et al. Regulatory cell therapy in kidney transplantation (The ONE Study): A harmonised design and analysis of seven non-randomised, single-arm, phase 1/2A trials. Lancet 2020, 395, 1627–1639.
- Peckert-Maier, K.; Schonberg, A.; Wild, A.B.; Royzman, D.; Braun, G.; Stich, L.; Hadrian, K.; Tripal, P.; Cursiefen, C.; Steinkasserer, A.; et al. Pre-incubation of corneal donor tissue with sCD83 improves graft survival via the induction of alternatively activated macrophages and tolerogenic dendritic cells. Am. J. Transplant. 2021.